【連載】よくわかるBDNF -基礎から臨床まで-「第4回 BDNF/TrkB シグナル もう一つの顔」
本記事は、和光純薬時報 Vol.90 No.2(2022年4月号)において、昭和大学 医学部 安達 直樹様に執筆いただいたものです。
はじめに
シリーズ第3回までで見てきたように、BDNF/TrkBシグナルは、神経回路の正常な発達や神経伝達調節において有益な機能を果たす。そのため、BDNF/TrkBシグナルの障害は脳の疾患の原因ともなり、その賦活化をターゲットとした創薬研究も数多くなされている。しかし今回は、そんなBDNF/TrkBの機能が、私たちの健康にとって「敵」となりうる事例について見ていきたい。
最初に主要なBDNF/TrkBシグナルについてもう一度確認をして、個別の疾患におけるBDNF/TrkBシグナルの関わりを紹介する。
BDNF/TrkBシグナル経路
MAPK/ERK経路
TrkB受容体のTyr490とTyr515残基が活性化されると、これらのチロシン部位にShcアダプタータンパク質がドッキングし、Grb2(Growth factor receptor-bound protein 2)をリクルートすることで、GTPase Rasと複合体を形成し、次にMAPK(mitogen activated protein kinase)/ERK(extracellular signal regulated kinase)経路を活性化する。MAPK/ERKキナーゼは、転写因子CREB(cAMP response element binding protein)をリン酸化して活性化する。リン酸化されたCREBは核内に移動し、様々な遺伝子のプロモーターに結合して転写を調節することで、細胞の生存や分化、増殖を促進する。
PI3K/Akt経路
PI3(Phosphoinositide 3)キナーゼ経路の活性化は、Tyr515残基のRasの複合体を取り込み、PI3K/Akt経路とMEK/MAPK経路という複数のカスケードを活性化させる。PI3K/Aktカスケードの活性化は、神経細胞の生存、成長および分化に不可欠なタンパク質群を制御する。
PLCγ経路
TrkB受容体のTyr816残基がリン酸化されると、PLCγ(phospholipase Cγ)経路が活性化され、IP3、DAG(diacyl glycerol)が生成される。PLCγ/IP3経路は、小胞体からのカルシウム放出をもたらし、CaMKⅡ(Ca2+/calmodulin-dependent protein kinaseⅡ)を活性化し、これによりCREBリン酸化を活性化する。一方、DAGの生成は、PKC(Protein kinase C)を活性化する。CaMKII・PKCの活性化は、細胞の生存や神経突起伸長、シナプス可塑性を担う。
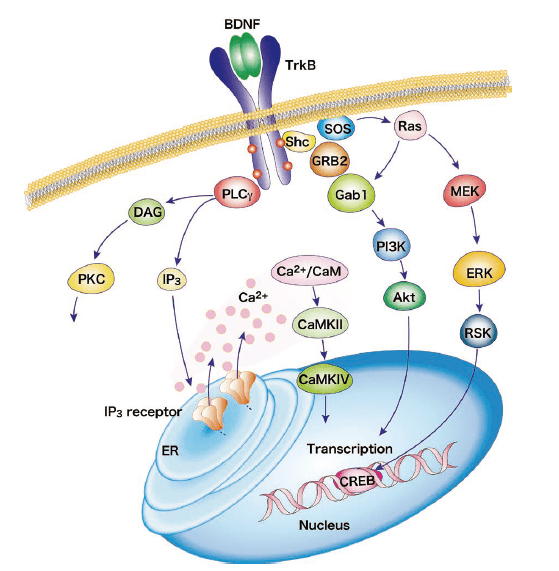
図.BDNF/TrkB シグナル経路 筆者ら文献37)より改変
薬物中毒における BDNF/TrkBシグナル
BDNF/TrkBシグナルは、脳の「報酬系」神経回路の可塑的な変化を促進することによって、コカイン中毒などの薬物依存症に関わる1)。報酬系を担う脳領域の基底状態において、BDNFは腹側被蓋野(VTA)、扁桃体、海馬、前頭皮質で高発現している一方で、背側線条体と側坐核(NAc)での発現量は低い2)。
これに対しTrkBは脳全体に広く発現している3)。齧歯類を用いた研究において、コカイン曝露は、これら報酬関連領域のBDNFレベルを上昇させることがわかっている4, 5)。また、コカイン中毒の動物モデルでは、VTAやNAcへのBDNF注入やBDNF/TrkBの強制発現によっては、条件付け場所嗜好性やコカイン探索行動、離脱症状が亢進し、抗BDNF抗体の注入やBDNF/TrkBのノックダウンによっては、これらの行動が抑制される6)。
コカインによるNAcなどにおけるBDNF/TrkBシグナルの増強が、AMPA型グルタミン酸受容体のサブユニットの構成を転写・翻訳レベルで調節することによってシナプスの可塑的変化を引き起こし、中毒症状の発現に関わるというモデルが示されている1)。一方、齧歯類の研究において、内側前頭前野皮質(mPFC)のBDNF/TrkBシグナル増強が、コカイン中毒症状において抑制的な役割を果たすことから7)、BDNF/TrkBシグナル抑制をターゲットとしたコカイン中毒治療は、NAcやVTA領域に限定したものであれば、十分に効果を発揮する可能性がある。
慢性疼痛における BDNF/TrkB シグナル
痛みの感覚刺激は、後根神経節ニューロンが一次求心性神経となり、脊髄後角に存在する二次ニューロンに興奮性の入力を送ることによって伝達されるが、この脊髄後角ニューロンの興奮性増大が、神経障害性疼痛の大きな要因となっていることが知られている。脊髄後角のBDNF/TrkBシグナルの増強が、神経障害性疼痛発症と進行に、以下の3つのメカニズムで、大きく寄与する可能性が指摘されている。
① KCC2
脊髄には、一次求心性神経のシナプス前抑制と後角投射ニューロンのシナプス後抑制という、2つの異なる抑制メカニズムが存在し、痛みの信号を制御している。BDNFはこの抑制性GABA作動性シナプスの調節に関与して痛覚過敏を引き起こす8)。ここで鍵となるのは、potassium chloride co-transporter 2 (KCC2)の機能である。KCC2は膜貫通型のタンパク質で、細胞内の塩化物イオン (Cl-) を細胞外に汲み出すことで、細胞外のCl-濃度を高め、GABAを受容したシナプス後細胞がGABA受容体開口時に過分極するという、GABAの抑制性を担保するポンプである9)。
神経障害性疼痛の動物モデルでは、脊髄後角においてBDNFの発現が上昇し、KCC2タンパク質の発現が低下する10)。さらに、TrkB/FcキメラまたはTrkB阻害剤K252aの髄腔内投与によって、末梢神経損傷による脊髄後角のKCC2発現低下がブロックされ、神経障害性疼痛が緩和される11)。まだ詳細はわかっていないが、BDNF/TrkBシグナルの中でも、Shc経路とPLCγ経路とが連動して活性化された場合に、KCC2の転写抑制が起こるようである12)。
②興奮性の増強
神経損傷モデルにおいて、BDNFによるTrkB受容体の活性化は、前シナプス部におけるグルタミン酸のシナプス小胞の蓄積を増やすことでシナプス伝達が増強することや、NMDA型受容体の機能を増強させることで、神経障害性疼痛を発症させる可能性も指摘されている13)。脊髄後角ニューロンのTrkB受容体にBDNFが結合すると、FynキナーゼによるGluN2B(NR2B)サブユニットのリン酸化を介して、後根神経節ニューロン−脊髄後角ニューロン間シナプスの長期増強を誘導し、中枢性感作と神経障害性疼痛を引き起こすと考えられている14, 15)。
③グリア細胞の活性化
上でBDNFによる脊髄後角ニューロンの興奮性増大が神経障害性疼痛を引き起こすメカニズムを見てきたが、神経損傷時に、最初にBDNFを供給するのはどの細胞であろうか? これまでの研究によって、BDNFの供給源がグリア細胞であることがわかってきている。
末梢神経損傷などによって活性化したミクログリアは、多くのサイトカイン(IL-2、TNF-α、BDNFなど)を産生・放出し、神経炎症反応と神経免疫応答の両方を誘導することで、疼痛過敏症の病態形成において重要な役割を果たす16)。活性化したミクログリア細胞では、BDNF産生が増加し、細胞移動が活発になり、TNF-α放出を増加する17)。さらに、外因性BDNFによってミクログリアが活性化し、細胞遊走とTNF-α放出を増加させることから、神経損傷時にはミクログリア由来のBDNFがオートクラインに作用して、さらに自身を活性化するという、正の循環ループ機構が存在する可能性がある18)。
また、脊髄後角のアストロサイトの活性化によっても、BDNFと炎症性サイトカインレベルが上昇するなど19)、アストロサイトも、持続的な中枢感作の主要な構成要素とみなされている20)。本来、BDNFの産生と放出は神経細胞を保護し、生存を促進するため、急性の危険因子(神経損傷など)への応答としてのBDNFの産生や放出増加は、生物として適応的であるはずなのだが、負のフィードバック機構が存在しないことが原因で、神経障害性疼痛の進行に伴って、中枢神経系が非適応状態へと変化しているのかもしれない。
てんかんにおける BDNF/TrkB シグナル
現在、世界の人口の約0.5 ~ 1%が罹患しているてんかんは、脳の神経細胞群が突然過活動する慢性進行性の中枢神経系障害である21)。てんかんは、自発的反復発作を特徴とし、一時的な脳機能障害や神経障害、認知機能低下など、さまざまな深刻な事態を引き起こす22)。
臨床研究において、てんかん患者の大半が健常者よりも血清中のBDNF値が高いことが知られている(Iughettiら、2018年)(Demirら、2020年)。また、てんかんの動物モデルでは、BDNFとTrkBが、特に側頭部と海馬領域で増加する23)。このようなマウスてんかんモデルにおいて、TrkBを欠失させたり24)、TrkB受容体を一過性に阻害したりすることで25)、てんかん発作の発生を著しく減少させることができる。さらに、ラットの海馬領域にBDNFを投与すると、てんかん発作を誘発しうる26)。
BDNF/TrkBシグナルは、どのようにてんかん発症にかかわるのだろうか。現在までに、主に3つのメカニズムが提唱されている。①BDNFはリアノジン受容体を介したCa2+放出と活性酸素種(ROS)産生を誘導し27)、神経細胞の損傷とアポトーシスを促進する28)。②TrkB活性化の上昇がKCC2の発現を低下させることで、GABAニューロンによる抑制効果を低下させる25)。③BDNF/TrkBシグナルの増強が、海馬歯状回顆粒細胞の軸索である苔状線維において異常発芽を起こし、脳内の異常興奮回路の形成を引き起こす29)。
BDNF/TrkBのてんかん発症における正確な役割は、BDNFの発現量、発現する脳部位、その上流および下流分子などとの関係から、まだまだ議論の余地があるが、BDNF/TrkBシグナル伝達経路に含まれる多くのタンパク質や分子は、臨床的なてんかんの検出や治療の分子標的として期待されており、てんかんの臨床治療やリスクの評価、予後予測に活用できる可能性がある。
がんにおける BDNF/TrkBシグナル
BDNF/TrkBシグナルは、細胞のがん化、浸潤や転移の促進、そして化学療法に対する抵抗性の原因となることが明らかにされている。
TrkBを過剰発現させた神経堤由来の細胞株をマウスに移植すると、10日後に急速に成長する腫瘍を形成し、腫瘍形成後7日以内にすべてのマウスが死亡した30)。また、脳腫瘍形成細胞は、TrkBを過剰に発現し、自身がBDNFを産生する限り、EGFなど本来の成長因子がなくても生存できることから、オートクラインで自身の生存を維持する能力を獲得していると考えられる31)。
腫瘍の治療を困難にする要因として、他の臓器への転移がある。腫瘍細胞からすると転移は自身の生き残りをかけたイベントである。細胞外基質から離れることで起こる細胞死(アノイキス)の壁を突破し32)、遺伝子発現を変化させることによって、上皮性から間葉性へと性質を変え(上皮―間葉転換)、血液を介して離れた臓器への移動に耐えられるようにしなければならない32, 33)。
興味深いことに、腫瘍細胞は、TrkBの活性化によってPI3キナーゼが常時活性化状態となることで、アノイキスに対する抵抗性を上げ、転移の成功率を高めるようだ32, 33)。本稿では詳細なメカニズムについては割愛するが、BDNF/TrkBシグナルが内因性・外因性のアポトーシスに対して抑制的に働くことで、抗がん剤化学療法に対する抵抗性を付与していると考えられている34)。
このように、BDNF/TrkBシグナルはがんの形成や転移、治療抵抗性に寄与する一方で、"有益"な機能も報告されている。メラノーマを移植したマウスは、環境エンリッチメント(遊び道具などがいくつもあるケージで飼育)の条件では、ナチュラルキラー細胞およびT細胞による細胞傷害性が増強され、腫瘍の成長が著しく抑制されることが見いだされた35, 36)。環境エンリッチメントマウスの視床下部ではBDNFの発現が増加しており、このBDNFをノックダウンすると、環境エンリッチメントによる腫瘍抵抗性が消失することから35)、視床下部におけるBDNF上昇が環境エンリッチメントによる抗腫瘍作用の鍵となっていると考えられている。
おわりに
本稿では、我々の健康にとって都合の悪い、BDNF/TrkBシグナルの「もう一つの顔」を見てきたが、基本的にはBDNFがTrkBに結合することによって、種々の細胞内シグナルカスケードが活性化され、その結果、シナプス伝達を調節したり、細胞の生存や増殖を促進したりする点では、有益な機能を果たす場合と変わらない。ただ、BDNF/TrkBシグナルが異所的に、もしくは過剰に活性化されることで、私たちの健康を脅かす存在にもなるのだ。
脳の疾患や機能障害に対する治療効果や、視床下部を介した抗がん作用への期待から、脳におけるBDNFの発現を上昇させることを目的として、AMPA型グルタミン酸受容体の反応を高めるアロステリックモジュレーター(ampakineと総称される)の開発が行われてきた。あるampakineでは、急性投与から数日以内に脳のいくつかの領域でBDNF産生を最大20倍まで増加させることが示されたが、意図しない発作原性作用が現れるなど、これらの薬剤の臨床応用には高い壁がある。しかし最近、発作性の副作用を示さず、加齢に伴う記憶障害を改善する最初のampakineが報告され36)、明るい光も見えてきている。
今回、負の側面に焦点を当てることで、BDNF/TrkBシグナルが適切な領域で適度に活性化されることの必要性と、臨床応用に向けた基礎的な研究の積み重ねの重要性を再認識できたかと思う。
参考文献
- Li, X. and Wolf, M. E. : Behav. Brain Res., 279, 240 (2015).
- Conner, J. M. et al. : J. Neurosci., 17, 2295 (1997).
- Krause, S. et al. : J. Neurosci. Res., 86, 411 (2008).
- Graham, D. L. et al. : Nat. Neurosci., 10, 1029 (2007).
- Horger, B. A. et al. : J. Neurosci., 19, 4110 (1999).
- Bahi, A. et al. : Psychopharmacology (Berl), 199, 169 (2008).
- Berglind, W. J. et al. : Eur. J. Neurosci., 26, 757 (2007).
- Malan, T. P. et al. : Anesthesiology, 96, 1161 (2002).
- Janssen, S. P. et al. : Neurochem. Int., 60, 21 (2012).
- Malcangio, M. : Neurobiol. Pain, 1, 1 (2017).
- Yajima, Y. et al. : J. Neurochem., 93, 584 (2005).
- Lee-Hotta, S. et al. : Neurochem. Int., 128, 32 (2019).
- Wang, L. N. et al. : J. Neurosci. Res., 90, 1249 (2012).
- Li, S. et al. : Neurochem. Res., 42, 2712 (2017).
- Ding, X. et al. : Neurobiol. Dis., 73, 428 (2015).
- Tsuda, M. et al. : Trends Neurosci., 28, 101 (2005).
- Zhang, X. et al. : Cell. Physiol. Biochem., 34, 715 (2014).
- Zhou, W. et al. : Neurosci. Lett., 756, 135965 (2021).
- Chiang, C. Y. et al. : Neurochem. Res., 37, 2419 (2012).
- Liu, T. et al. : Neurosci. Bull., 28, 131 (2012).
- Fisher, R. S. et al. : Epilepsia, 55, 475 (2014).
- Thijs, R. D. et al. : Lancet, 393, 689 (2019).
- Wang, X. et al. : Front. Pharmacol., 12, 758232 (2021).
- He, X. P. et al. : Neuron, 43, 31 (2004).
- Liu, G. et al. : Neuron, 79, 31 (2013).
- Scharfman, H. E. et al. : Exp. Neurol., 174, 201 (2002).
- Yang, N. et al. : Oxid. Med. Cell. Longev., 2020, 6687185 (2020).
- Ghadiri, T. et al. : Neurosci. Lett., 709, 134384 (2019).
- Skupien-Jaroszek, A. et al. : PLoS One, 16, e0239111 (2021).
- Yin, B. et al. : Oncogene, 34, 761 (2015).
- Lawn, S. et al. : J. Biol. Chem., 290, 3814 (2015).
- Bao, W. et al. : PLos One, 8, e70616 (2013).
- Kupferman, M. E. et al. : Oncogene, 29, 2047 (2010).
- Radin, D. P. and Patel, P. : Anticancer Res., 37, 3983 (2017).
- Cao, L. et al. : Cell, 142, 52 (2010).
- Radin, D. P. et al. : Biomed. Pharmacother., 84, 806 (2016).
- Numakawa, T. et al. : World J. Biol. Chem., 1, 133 (2010).



