【連載】よくわかるBDNF -基礎から臨床まで-「第3回 BDNFと精神疾患との関与(BDNF経路を介した可塑性の制御と精神疾患)」
本記事は、和光純薬時報 Vol.90 No.1(2022年1月号)において、ヘルシンキ大学 ニューロサイエンスセンター 梅森 十三様に執筆いただいたものです。
はじめに
これまでに、BDNF経路は神経可塑性の制御に関わっていることを述べた。可塑性が高まると、学習や環境の変化に適応して、神経回路は構造的、機能的に再編成される。病態生理学的な(前)臨床研究により、統合失調症やうつ病などの精神疾患において、BDNFおよびその受容体であるTrkBを介したシグナル伝達の異常が報告されている。また近年、抗うつ薬がBDNF/TrkBシグナル伝達を通じた神経可塑性を調節するメカニズムが解明されている。今回はBDNF/TrkB経路の異常と精神疾患の関係、加えてこの経路を標的とした神経精神疾患様の症状の改善について、最新の研究結果を交えながら解説する。
統合失調症における BDNF/TrkB経路の異常
BDNF経路は、胎生期や臨界期の脳の発達だけではなく、成体においても海馬や大脳皮質神経細胞の可塑性や生存の制御に重要な役割を持っている。したがって、BDNF経路の異常が統合失調症患者の脳の変化に関わっている可能性は想像に難くない。実際に統合失調症のモデル動物ではBDNFの調節異常が見られ、TrkBの前脳特異的ノックアウトマウスでは運動亢進や常同行動、認知障害などの統合失調症様の行動が見受けられる。
また、統合失調症患者の死後脳の解析では、大脳皮質領域と海馬においてBDNF濃度の減少が見られるが前部帯状皮質では上昇していることが報告されている。これらのBDNFの発現異常が、統合失調症患者の神経発達と神経可塑性の異常をもたらしている可能性が示唆されている1-3)。
気分障害におけるBDNF/TrkBシグナル伝達異常
BDNFのメッセンジャーRNAとタンパクは、自殺者や大うつ病性障害(うつ病)患者の死後脳、特に海馬と扁桃体で減少していることがわかっている4)。また、うつ病患者や自殺者の末梢血単核細胞において、BDNF遺伝子プロモーターのDNAメチル化が増加することも示されており、BDNF発現の調節不全が示唆されている5)。
さらに、第2回でも説明した通り、ヒト血清では血小板由来と考えられるBDNFが高いレベルで検出される。しかし興味深いことに、うつ病患者の血清または血漿BDNFレベルが低く、抗うつ薬による治療が成功した後に元のレベルに戻ることが観察されている。同様に血清BDNFについても、未治療のうつ病患者は、治療に成功した患者や健常群と比較して、有意な減少が見られている4, 6)。
また、血清のBDNFとproBDNFの比率は健常群に比べて双極性障害では高く、うつ病では低くなっていることから、両者を区別できるマーカーになると期待されている7, 8)。ただしこれらのBDNFレベルは、上述の通り統合失調症1, 3)で低下しており、自閉症では上昇と低下の両方が報告されている9)。また、血清BDNF含量は個体間および個体内でのばらつきが大きいことから、信頼性や疾患特異性についてはさらなる検討が必要と考えられる。
BDNFに加えて、TrkBのタンパクおよびmRNAのレベルもうつ病患者の死後脳で減少し、TrkBの遺伝子変異は自殺に関連していることが報告されている。さらに、活性化状態であるリン酸化TrkBは、うつ病患者の脳サンプルで減少することがわかっている4)。
BDNFと精神疾患の遺伝的関連
BNDFの66番目のアミノ酸残基はバリンが大部分であるが、Caucasoidでは25%から50%がこの位置にメチオニンを持っている(Val66Met多型)10, 11)。この多型はうつ病や統合失調症などの精神疾患だけでなく、多発性硬化症、アルツハイマー病、パーキンソン病などの神経変性疾患の危険因子である可能性が示唆されている12, 13)。
うつ病においては、この多型は幼少期の虐待や貧困、成人期の慢性ストレスを原因とするうつ病への発症しやすさに関与しているとの報告がある14)。また動物実験においては、この多型は空間記憶や不安様行動に影響し、細胞内BDNF輸送と神経活動依存的BDNF放出に影響を及ぼすこと10, 15)、BDNF mRNAの樹状突起輸送を損なうこと16)、さらには長期抑圧(long-term depression : LTD)を低下させること17)が示されている。つまり、この多型はBDNFの活動依存的な神経可塑性に影響している可能性が示唆されている。
抗うつ薬による BDNF/TrkB シグナルを介した可塑性の上昇
BDNF/TrkBシグナル伝達がうつ病患者の血中と脳内で減少していることを考えると、抗うつ薬がBDNFレベルを増加させるという発見は大変興味深いものであった。実際に動物実験によって、三環系モノアミン酸化酵素阻害薬および選択的セロトニン再取り込み阻害薬(Serotonin selective reuptake inhibitor : SSRI)の抗うつ薬が、脳内のBDNF発現を増加させることがわかった6)。
また、マウス海馬の歯状回細胞からのBDNFや歯状回顆粒ニューロンの前駆細胞からのTrkB発現の欠失は、うつ様行動および神経新生に対する抗うつ薬の効果を減少させることが示された6)。近年、即効性の抗うつ薬として注目されている麻酔薬ケタミン(NMDA型グルタミン酸受容体の拮抗薬)においても、マウス前脳領域におけるBDNFまたはTrkB遺伝子の欠失により、うつ様行動の減少や海馬シナプス増強などの抗うつ作用が抑えられる18)。
また興味深いことに、抗うつ薬に対する行動および神経可塑性に関連する反応は、Val66Met多型のマウスモデルで失われていた15, 19)。さらに、これらのマウスでは、ケタミンとその代謝物である(2R、6R)-ヒドロキシノルケタミン(hydroxynorketamine ; HNK)によって誘導されるスパイン形成の増加が抑えられていた18, 20)。HNKはケタミンの抗うつ作用を主に担っており、BDNF/TrkBシグナル伝達を通した神経可塑性の変化をもたらすと考えられている21)。
しかし、臨床研究においては、Met対立遺伝子はどちらかといえば従来の抗うつ薬への反応を改善していることも示唆されている22-25)。一方反応の減少も報告された26)。うつ病患者の抗うつ反応におけるBDNFシグナル伝達の役割を解明するには、さらに多くの遺伝的解析が必要と考えられる4)。
若年期可塑性の誘導(iPlasticity)による神経精神疾患様の症状の改善
筆者が所属するヘルシンキ大学、ニューロトロフィン研究室(Eero Castren教授)では、抗うつ薬の慢性投与によって誘導される、幼弱期や臨界期に見られるような神経可塑性(induced juvenile critical period-like plasticity : iPlasticity)に焦点を当てて研究を行っている。Castrenは、この可塑性の上昇は、ある種の「トレーニング」を組み合わせることにより、うつだけではなく他の神経精神疾患の治療にも応用できる可能性を提唱している27)。
つまり代表的なSSRIであるフルオキセチンをラットあるいはマウスに慢性投与した後に、単眼遮蔽を組み合わせることで弱視を、恐怖記憶消去トレーニングにより心的外傷後ストレス障害(PTSD)を、さらには社会性の訓練により攻撃性が改善されることを明らかにした28-31)。これはフルオキセチンがBDNF/TrkBの活性化を介した神経可塑性の上昇に加えて、内外刺激により神経が活性化されて、脳神経ネットワークの再編成を引き起こすためであると考えられている。
しかし、抗うつ薬がどのようにTrkBを活性化するのか、またどの神経ネットワークがどのような変化を受けるのかが不明だった。最近、フルオキセチン、イミプラミン、ケタミンなど、さまざまなクラスに属する抗うつ薬が直接TrkBに結合することが示された32)。二量体を形成したTrkBは膜貫通ドメインで互いに交差し、抗うつ薬が結合するためのポケットを作成する。抗うつ薬は、このポケット内のコレステロール結合ドメインに直接結合し、シナプス膜のTrkBを安定化し、BDNFを介したTrkBシグナル伝達を促進する32)。これらの発見は、抗うつ薬の主要な作用部位がモノアミン輸送体ではなく、TrkBへの直接結合であるという全く新しい仮説を提唱し大きな話題になっている。
さらに我々は、光刺激によって二量体化するoptoTrkBの系を用いることにより、これまでBDNFは拡散性であるが故に難しかった標的細胞の可塑性の研究を行っている。パルブアルブミン陽性介在神経細胞でTrkBの活性化と単眼遮蔽を組み合わせることで、眼優位性が引き起こされ、シナプス可塑性の上昇、脱抑制によるLTPの上昇や脳波の同調をもたらすことを証明した33)。現在はoptoTrkBの系を用いて、PTSDの治療や空間記憶の 向上、薬物依存性の軽減を目指して研究を行っている。
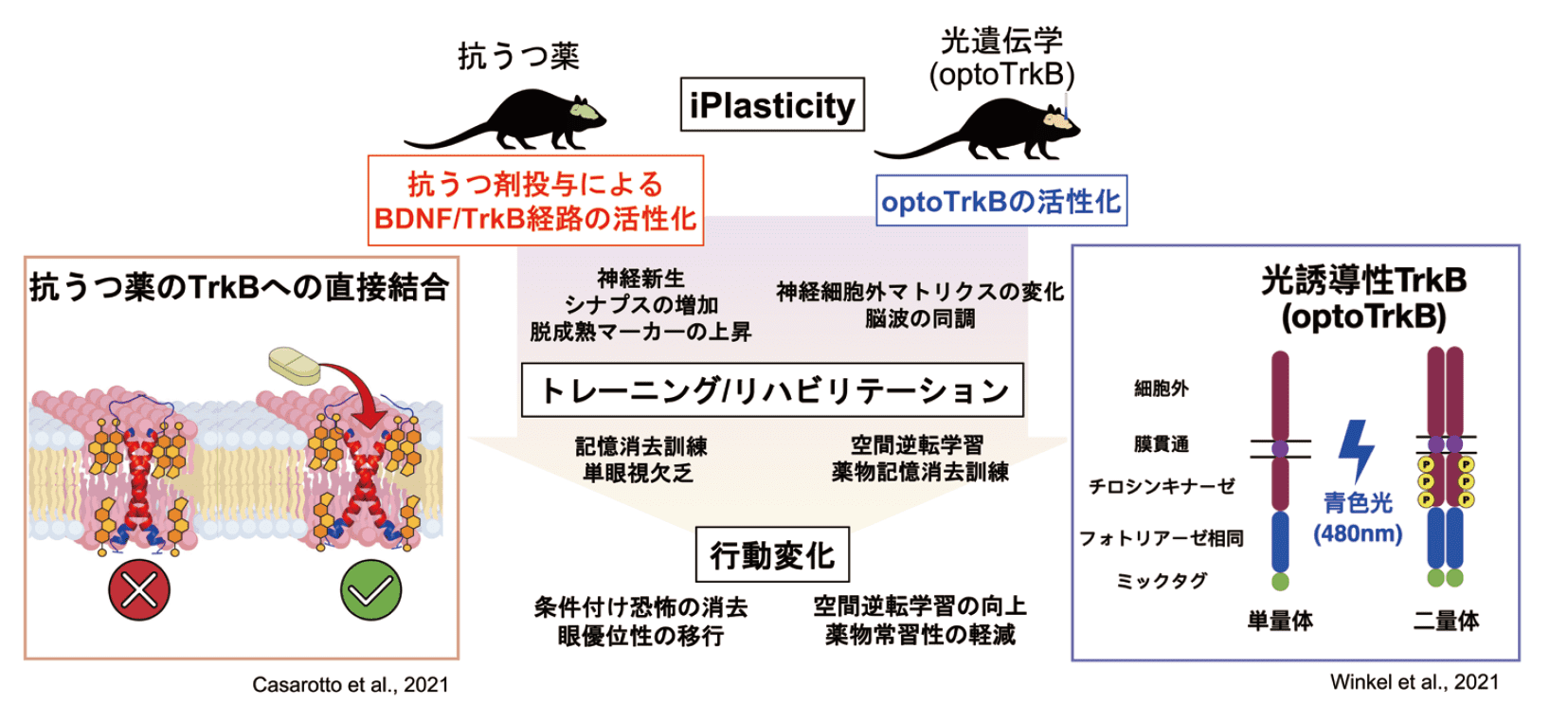
図.若年期可塑性の誘導(iPlasticity)による神経精神疾患様の症状の改善
おわりに
Val66Met多型の発見以降、多くの遺伝研究によりその遺伝的影響や抗うつ薬の効果の違いを生み出す可能性が示されてきた。その一方、いくつかの研究ではその効果を再現することができていない。これは年齢や性別、環境要因、民族性、分析に使用される遺伝子モデル、遺伝子間相互作用などの多くの要因が複雑に影響しあっているためであると考えられる34)。動物モデルを使った研究では、Val66Met多型モデルマウスや抗うつ薬作用の新しいモデル、optoTrkBを用いることによって、BDNF経路が関与する精神疾患の発症メカニズムの解明や新たな治療法を見出す可能性が期待される。
謝辞
藤田医科大学 小清水先生には有益な助言をいただきました。この場を借りてお礼申し上げます。
参考文献
- Angelucci, F. et al. : Mol. Psychiatry, 10(4), 345 (2005). DOI: 10.1038/sj.mp.4001637
- Fernandes, B. S. et al. : Mol. Psychiatry, 20, 1108 (2015). DOI: 10.1038/mp.2014.117
- Nieto, R. R. et al. : Front. Psychiatry, 12, 662407 (2021). DOI: 10.3389/fpsyt.2021.662407
- Castrén, E. and Monteggia, L. M. : Biol. Psychiatry, 90, 128 (2021). DOI: 10.1016/j.biopsych.2021.05.008
- Hing, B. et al. : Am. J. Med. Genet. B Neuropsychiatr. Genet., 177, 143 (2018). DOI: 10.1002/ajmg.b.32616
- Autry, A. E. and Monteggia, L. M. : Pharmacol. Rev., 64, 238 (2012). DOI: 10.1124/pr.111.005108
- Yoshida, T. et al. : PLoS One, 7, e42676 (2012). DOI: 10.1371/journal.pone.0042676
- Södersten, K. et al. : J. Affect. Disord., 160, 1 (2014). DOI: 10.1016/j.jad.2014.01.009
- Saghazadeh, A. and Rezaei, N. : J. Autism Dev. Disord., 47, 1018 (2017). DOI: 10.1007/s10803-016-3024-x
- Egan, M. F. et al. : Cell, 112, 257 (2003). DOI: 10.1016/s0092-8674(03)00035-7
- Shimizu, E. et al. : Am. J. Med. Genet. B Neuropsychiatr. Genet., 126B, 122 (2004). DOI: 10.1002/ajmg.b.20118
- Bath, K. G. and Lee, F. S. : Cogn. Affect. Behav. Neurosci., 6, 79 (2006). DOI: 10.3758/CABN.6.1.79
- Shen, T. et al. : Aging Dis., 9, 523 (2018). DOI: 10.14336/AD.2017.0717
- Hosang, G. M. et al. : BMC Med., 12 : 7, 1 (2014). DOI: 10.1186/1741-7015-12-7
- Chen, Z.-Y. et al. : Science, 314, 140 (2006). doi: 10.1126/science.1129663
- Baj, G. et al. : Front. Neurosci., 7, 188 (2013). DOI: 10.3389/fnins.2013.00188
- Mizui, T. et al. : Proc. Natl. Acad. Sci. U. S. A., 112, E3067 (2015). DOI: 10.1073/pnas.1422336112
- Björkholm, C. and Monteggia, L. M. : Neuropharmacology, 102, 72 (2016). DOI: 10.1016/j.neuropharm.2015.10.034
- Bath, K. G. et al. : Neuropsychopharmacology, 37, 1297 (2012). DOI: 10.1038/npp.2011.318
- Fukumoto, K. et al. : Proc. Natl. Acad. Sci . U. S. A., 116, 297 (2019). DOI: 10.1073/pnas.1814709116
- Zanos, P. et al. : Nature, 533, 481 (2016). DOI: 10.1038/nature17998
- Choi, M. J. et al. : Brain Res., 1118, 176 (2006). DOI: 10.1016/j.brainres.2006.08.012
- Niitsu, T. et al. : Prog. Neuro-psychopharmacol. Biol. Psychiatry, 45, 183 (2013). DOI: 10.1016/j.pnpbp.2013.05.011
- Yan, T. et al. : Asia-Pac. Psychiatry, 6, 241 (2014). DOI: 10.1111/appy.12148
- Domschke, K. et al. : Int. J. Neuropsychopharmacol., 13, 93 (2010). DOI: 10.1017/S1461145709000030
- Laje, G. et al. : Biol. Psychiatry, 72, e27 (2012). DOI: 10.1016/j.biopsych.2012.05.031
- Castrén, E. : Nat. Rev. Neurosci., 6, 241 (2005). DOI: 10.1038/nrn1629
- Umemori, J. et al. : Psychiatry Clin. Neurosci., 72, 633 (2018). DOI: 10.1111/pcn.12683
- Mikics, É. et al. : Neuropsychopharmacology, 43, 235 (2017). DOI: 10.1038/npp.2017.142
- Karpova, N. N. et al. : Science, 334, 1731 (2011). DOI: 10.1126/science.1214592
- Vetencourt, J. F. M. et al. : Science, 320, 385 (2008). DOI: 10.1126/science.1150516
- Casarotto, P. C. et al. : Cell (2021). doi: 10.1016/j.cell.2021.01.034.
- Winkel, F. et al. : Mol. Psychiatry, 1-10 (2021). doi: 10.1038/s41380-021-01211-0.
- Tsai, S.-J. et al. : Front. Mol. Neurosci., 11, 156 (2018). doi: 10.3389/fnmol.2018.00156



