【連載】遺伝子解析 新技術とその応用 「第4回 トランスクリプトーム・エピゲノムの統合解析」
本記事は、和光純薬時報 Vol.89 No.2(2021年4月号)において、熊本大学発生医学研究所 細胞医学分野 古賀 友紹様、中尾 光善様に執筆いただいたものです。

エピゲノムは、DNA のメチル化、ヒストンの化学修飾、クロマチンの高次形成、非コードRNA による調節などで構成されて、転写関連因子と協働することで、ゲノム上の全遺伝子発現が制御されると考えられる。エピゲノムには安定性と可逆性という両面があり、特定の遺伝子座を取り上げてもエピゲノム形成の特異性はほとんど未知である。近年のシークエンス技術の革新により、ゲノムワイドなエピゲノム解析が急速に発展している。多様なエピゲノムを網羅的に検討し、トランスクリプトームのデータと合わせることで、表裏一体の遺伝情報を検証することが可能になった。本稿では、高速シーケンサーを用いた3種類の基本技術 (RNA-seq、ATAC-seq、ChIPseq)について、実例を交えながら概説したい。
1. RNA-seq
RNA シーケンス (RNA-seq) は、RNA からcDNA ライブラリを作製し、高速シーケンサーを用いて取得したリード情報をもとにして、遺伝子の発現量を解析する手法である。イルミナ社やサーモフィッシャーサイエンティフィック社などから解析機器が提供されており、研究内容に合わせて選択する必要がある。RNA-seq では、poly Aを利用してmRNA を回収mRNA-seqを行ったり、rRNA を除去してwhole RNA-seq を行ったり、miRNA の網羅的解析を行ったりと、用途は様々である。
例えば我々は、ホルモン療法耐性化モデルの乳がん細胞株MCF7 を用いて、mRNA-seq とwhole RNA-seqの比較解析によって、エストロゲン受容体をコードするESR1 遺伝子座から新規の非コードRNA 群(Eleanors と名付けた)を同定した(図1 ; Tomita, S. et al. : Nat. Commun., 6, 6966 (2015). ; Abdalla, M. O. A. et al. : Nat. Commun., 10, 3778 (2019).)。MCF7細胞がエストロゲン枯渇に適応する過程で、ESR1 遺伝子座の活性化にEleanors が重要な役割を果たすことを明らかにした。
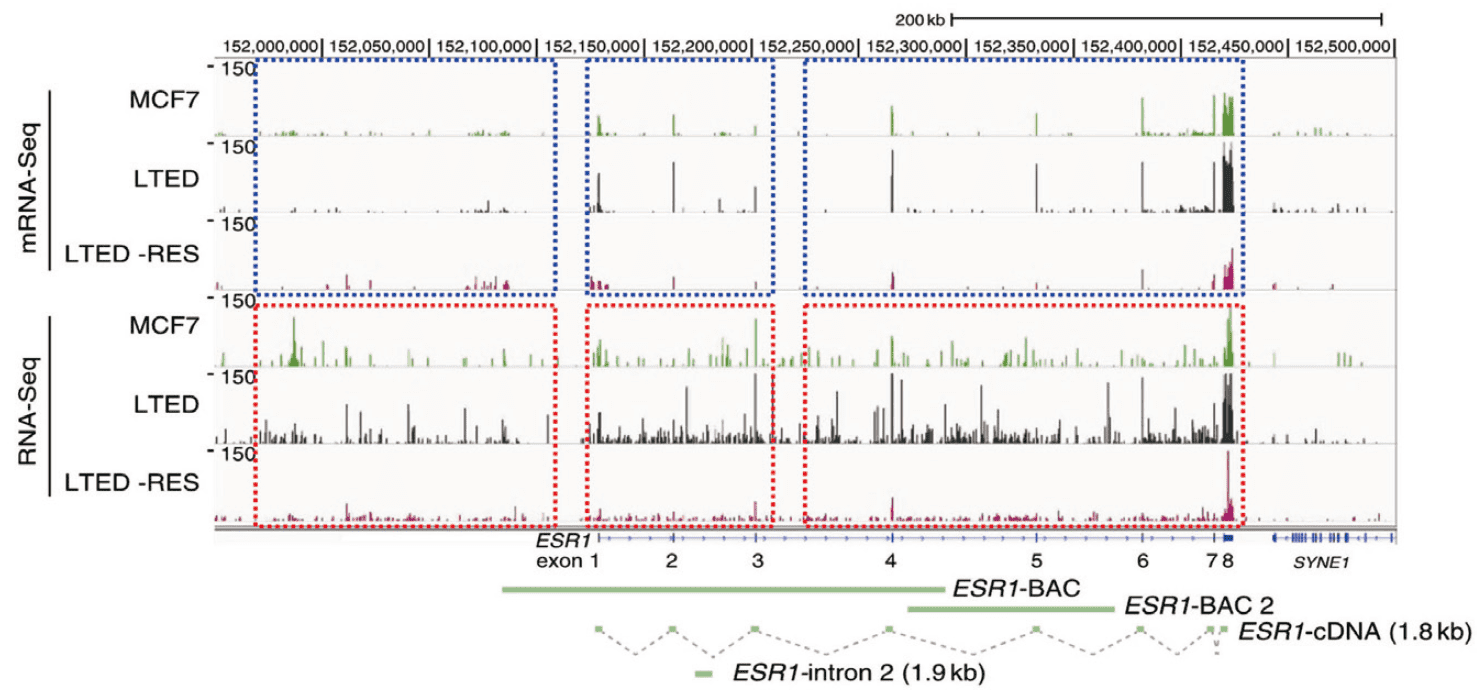
図1.エストロゲン受容体遺伝子座におけるmRNA-seqとwhole RNA-seqの比較( Tomita, S. et al. : Nat. Commun., 6, 6966 (2015).より改変。)
mRNA-seq ではexon 部位にそのピークが集積している(青枠)。Whole RNA-seq ではintron やpromoter、intergenic 部位にもピークが見られる(赤枠)。ピークの高さから親株のMCF7 よりもLTED で非コードRNA の発現が高いことがわかる。MCF7 : ヒト乳癌細胞株。LTED : MCF7を長期間エストロゲン欠乏培地で培養し、エストロゲン枯渇に適応した細胞株。LTED-RES : LTED にエストロゲン様作用のあるポリフェノールであるレスベラトロールを処理した状態。また近年では、急速な分析技術の高度化により、1個の細胞を分離・標識・解析できるようになった(シングルセルRNA-seq、scRNA-seq)。この手法では、細胞集団の転写産物を1 細胞ごとに網羅的な解析をするため、細胞集団の不均一性を解析するのに適しており、細胞集団を亜集団にクラスタリングしてそれぞれの集団の特徴遺伝子を抽出することができる。最大の利点は、集団の平均値を見る通常のバルクRNA-seq では検出が困難な希少細胞における特徴的な発現変化が、scRNA-seq では感度良く捉えられることである。近年では、データベースも充実してきており、Tabula-Muris などでは、20 のマウス臓器におけるscRNA-seq の全データを公開している(https://tabulamuris.ds.czbiohub.org)。
2. ChIP-seq
ChIP-seq とは、クロマチン免疫沈降 (chromatin immunoprecipitation: ChIP)と高速シーケンサーを組み合わせて、メチル化やアセチル化などの修飾型ヒストン、ヒストン修飾因子、クロマチンリモデラー、転写因子(DNA 結合タンパク質)などの、ゲノム上での結合部位を網羅的に解析する手法である。現在、エピゲノム解析を行うための基本的な方法になっている。クロスリンクしたDNA を断片化してChIP し、脱クロスリンク、DNA抽出を行い、ライブラリー調製後、シーケンスをゲノムワイドに解読する。ChIP-seq には特異的な抗体が免疫沈降に必要なため、標的とするタンパク質もしくはヒストン修飾などが決まっていること、また、ChIP に適した抗体があることが必須になる。
ChIP-seq では、ゲノムワイドなDNA-タンパク質相互作用について詳細な情報が得られるが、十分な量のDNAライブラリー調製が必要なため、大量の細胞が必要になる(通常、1 x 106個程度)。最近では、少量の細胞からChIP-seq を可能とする手法も開発されており、ヒストン修飾(H3K4me3、H3K27me3、H3K4me1) は5 x 103 個程度の細胞数があれば解析可能になっている。
また、転写因子などDNA に直接結合するタンパク質は比較的感度の良いデータが得られやすいが、ヒストン修飾酵素など、間接的にDNA に集積する因子はタンパク質間のダブルクロスリンクによる固定を用いるなど、予めの条件検討が必要である(Tanaka, H. et al. : Aging Cell, 19, e13173 (2020). ; Anan, K. et al. : Nucleic Acids Res., 46, 5441 (2018).)。またChIP-Atlas は、公開されている既報のChIP-seq データを再解析、またはユーザーデータを照合するウェブツールとして有用性が高い (https://chipatlas.org)。
3. ATAC-seq
Assay for Transposase-Accessible Chromatin (ATAC)-seq は、高活性改変型Tn5 transposase を用いて、DNA の断片化とタグ付けを行い、ゲノムワイドなクロマチン構造を解析するもので、2013 年に開発された(Buenrostro, J. D. et al. : Nat. Methods, 10, 1213 (2013).)。アッセイ自体の簡便さと幅広いサンプルに適用できることから、オープンクロマチン領域の同定や転写因子の占有率を評価するために有効である(図2)。
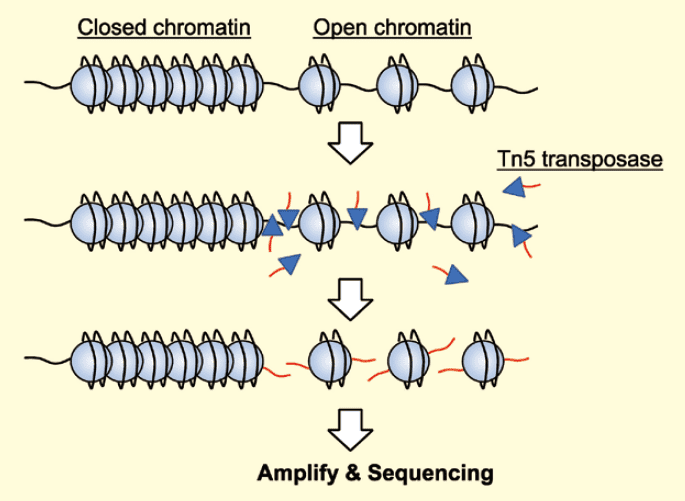
図2.ATAC-seq の原理
Tn5 transposase がアクセスできるオープンクロマチン領域が切断され、オリゴDNA が付加される。その後、DNA を抽出し、高速シーケンサーで解析する。個別のヒストン修飾の検討など、詳細なエピゲノム解析の前段階にも位置づけられる。同様の目的でこれまでに用いられていたDNase-seq やFormaldehyde-Assisted Isolation of Regulatory Elements(FAIRE)-seq が1 x 106 個以上の細胞を必要とするのに比べて、必要な細胞数が5 x 102 〜104 個と大幅に少ないことも利点である。
また近年では、Tn5 transposase に組み込むオリゴDNAを蛍光色素で標識しクロマチンと反応させることで、オープンクロマチン領域を蛍光標識し1細胞レベルで可視化するATAC-see という方法が開発されている(Chen, X. et al. : Nat. Methods, 13, 1013 (2016).)。この手法では、蛍光強度の高い細胞 (= オープンクロマチン領域が多い細胞)をフローサイトメトリーで分取した後、高速シーケンサーを用いてオープンなクロマチン領域を同定することができる。
ATACsee法ではフローサイトメトリーで解析できるため、異なる蛍光色素で標識した抗体と組み合わせることで、特定の細胞集団におけるクロマチン状態を解析するなど、応用範囲は広い。さらに、シングルセルATAC-seq(scATAC-seq)を用いると、単一細胞解像度でオープンクロマチン領域を評価できるため、scRNA-seq と組み合わせたゲノムワイドなシングルセル解析が加速している。
4. おわりに
エピゲノム解析の広さと深さは拡大し、生命現象の分子基盤を理解するために不可欠な技術に位置づけられる。今回紹介したRNA-seq、ChIP-seq、ATAC-seq に加えて、エピゲノムの構造・機能的な相互作用(染色体コンフォメーションキャプチャ 3C/4C/HiC-seq、HiC とChIP を組み合わせたHiChIP)なども含めると、様々なアプローチが可能である。バイオ情報の解析ツールも増えているので、これらのデータを照合して、統合的に理解することが重要であると考えられる。
−シリーズ終了にあたって−
2020 年07 月(Vol. 88 No. 3)から始まりました「遺伝子解析 新技術とその応用」は、本稿をもちまして最終回を迎えました。本誌に寄稿いただきました先生方にこの場を借りて御礼申し上げます。おかげさまで本シリーズでは遺伝子解析に関する情報をさまざまな視点でお届けできました。
読者の皆様におかれましては、全4回に渡って続いた本連載をご愛読いただき御礼申し上げます。皆様の遺伝子研究の一助になりましたら幸いです。



