【総説】家族性パーキンソン病患者iPS細胞由来神経系細胞におけるエンドソーム機能障害
本記事は、和光純薬時報 Vol.89 No.3(2021年7月号)において、東京慈恵会医科大学 再生医学研究部a) 脳神経内科b) 坊野 恵子様a),b)、岡野 ジェイムス 洋尚様a)に執筆いただいたものです。
はじめに
近年、不治の病とされた様々な神経疾患の原因遺伝子・感受性遺伝子が同定され、その根幹病態にせまる発見が相次ぎ、病態解明に向け新たな知見が蓄積してきている。疾患関連遺伝子から推測される病態経路を疾患モデル細胞やモデル動物を用いて検証することにより、新規治療法の研究・開発が飛躍的に進んできた。2006年に誕生したiPS細胞は、再生医療に重要な役割を果たすと期待されており、臨床応用に向けた研究が進んでいる。また、難治性疾患患者の体細胞からiPS細胞を作製し患部の細胞に分化させ病態を解明する研究も世界中で盛んに行われている。多分化能を維持しつつ無限に増殖するiPS細胞は、病態研究や新規薬剤探索など基礎・臨床橋渡し研究のリソースとしても極めて有用である。
iPS 細胞を用いたパーキンソン病の研究
パーキンソン病は、振戦、筋固縮、無動、姿勢反射障害の四大運動症状を特徴とする難病である1)。神経変性疾患ではアルツハイマー型認知症に次ぎ多く、高齢化に伴い患者数は増加している。パーキンソン病はニューロンにおけるレヴィ小体(α-シヌクレインが異常蓄積した凝集体)の出現を病理学的特徴とする2)。
大部分のパーキンソン病は孤発性であり、遺伝的要因と環境因子の複雑な組み合わせにより発症すると考えられているが、約10%は単一遺伝子異常により発症する家族性であることがわかっている。これまでに家族性パーキンソン病の原因遺伝子は22種類が報告されているが、これらの遺伝子の強制発現あるいはノックダウン培養株化細胞、モデル動物の解析から、ミトコンドリア障害、酸化ストレスによる障害、ユビキチン・プロテアソーム系やオートファジー系などタンパク質分解系の異常が病態に深く関与することが示されている3)。家族性および孤発性パーキンソン病に共通する病態メカニズムが存在すると推測されているため、家族性パーキンソン病の病態解析により、孤発性パーキンソン病の新規治療法開発に資する重要な情報が得られる可能性がある。
しかし、株化培養細胞モデルや動物モデルは種差や細胞特異性の違いが問題となることがあり、必ずしも病態を正確に反映する訳ではない。また、過剰発現系による疾患モデルでの研究結果を臨床試験の根拠として使用する場合、ヒトの生理的遺伝子発現量を反映していないため、試験の成功率を下げる潜在リスクとなることも指摘されてきた。そのため、これまでにパーキンソン病の病態進行の制御を目指した様々な治療戦略が検討されたにもかかわらず、根本的治療法は上市されてこなかった4)。
現在、iPS細胞技術の進歩により実際の患者の脳内で起きていると考えられる現象をin vitroで再現することが可能となった5)。特に非侵襲的に罹患組織から細胞を採取することが困難な神経変性疾患などの研究において、これまで不可能だったヒトニューロンの生細胞解析を行うことができる。さらに疾患特異的iPS細胞には、患者の遺伝的背景を保持しつつ目的の細胞に誘導できること、生理的な遺伝子発現量における解析が可能であること、ヒト細胞特異的な薬効評価が出来ることなどのアドバンテージがあり、病態解析や創薬研究のプラットフォームとして利用価値が高い。
VPS35 とパーキンソン病
PARK17は17番目に発見された家族性パーキンソン病であり、2011年にスイスとオーストリアからPARK17の家系が最初に報告された6, 7)。PARK17症例は日本でも報告されており、臨床症状は孤発性パーキンソン病と類似している8)。常染色体優性遺伝形式で遺伝するPARK17の原因遺伝子はVPS35であり、レトロマーのサブユニットタンパク質をコードしている9)。レトロマーは、VPS26-VPS29-VPS35からなる三量体と、sorting nexin二量体が構成するタンパク質複合体である10)(図1参照)。
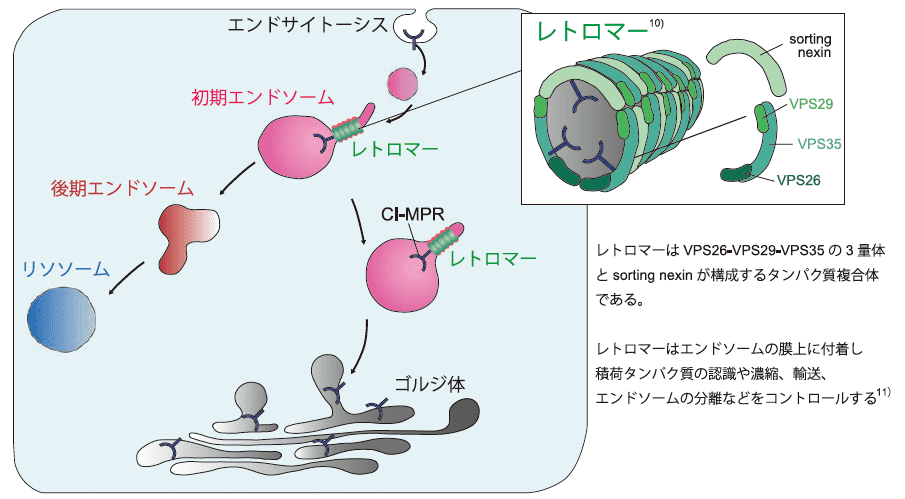
図1.
レトロマーはエンドソームの膜上に付着し、積荷タンパク質の認識、輸送、エンドソームの切断制御など、エンドソーム輸送機構の重要な役割を担っていることが近年明らかになった11)。パーキンソン病をはじめ病因タンパク質の細胞内蓄積が亢進する疾患では、異常タンパク質の細胞内輸送障害が病態に関わっていることが予測されていた12)。しかしながら、VPS35の変異がパーキンソン病の病態にどのようなメカニズムで関与しているのかは不明である。我々はVPS35(D620N)変異のPARK17患者さんのご協力のもと、末梢血単核球からiPS細胞を樹立し、神経細胞に分化させ解析を行った。
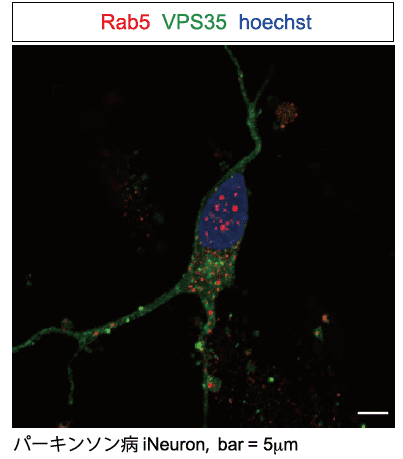
図2.
パーキンソン病は運動障害としてパーキンソニズムを呈し、その責任病巣である中脳黒質のドパミンニューロンの脱落が原因で発症する。そこで、疾患特異的iPS細胞からドパミンニューロンを分化誘導したところ、コントロールと比較して疾患群ではアポトーシスが誘導されやすく、特異的にドパミンニューロンの細胞数が減少することがわかった。
エンドソームは細胞内へと取り込まれた物質の選別、分解、再利用などを制御するオルガネラであり、細胞内の様々なタンパク質などを輸送する13)。受容体などの積荷タンパク質はエンドサイトーシスにより細胞表面から取り込まれ、膜で区切られた様々な大きさの球形の小胞(エンドソーム)によって輸送される。積荷タンパク質はまず初期エンドソームに入り、一部は再利用のためゴルジ体に逆行輸送され、一部は後期エンドソームを経てリソソームへと輸送され分解される(図1)。
そこで、iPS細胞から分化誘導したニューロン(iNeuron)を固定し、レトロマーを構成するVPS35とエンドソームを免疫染色し観察したところ、Rab5(初期エンドソームのマーカー)およびRab7(後期エンドソームのマーカー)の多くはVPS35と隣りあい、もしくは重なりあって局在しており、コントロール群とパーキンソン病群の間でその分布に差は認められなかった(図2)。
次に、エンドソーム輸送機構におけるVPS35(D620N)変異の影響を生細胞で調べるため、蛍光タンパク質と融合させたRab5aもしくはRab7aをiNeuronで発現させライブイメージングで観察した。Rab5aもしくはRab7a陽性小胞の細胞内動態を追跡した結果、疾患群では初期および後期エンドソームの移動速度がともに低下していた(図3)。さらに小胞の分離および融合の頻度も低下していることがわかった(図4)。この結果から、VPS35(D620N)変異が初期および後期エンドソームの輸送、分離、融合を阻害していることが明らかになった。
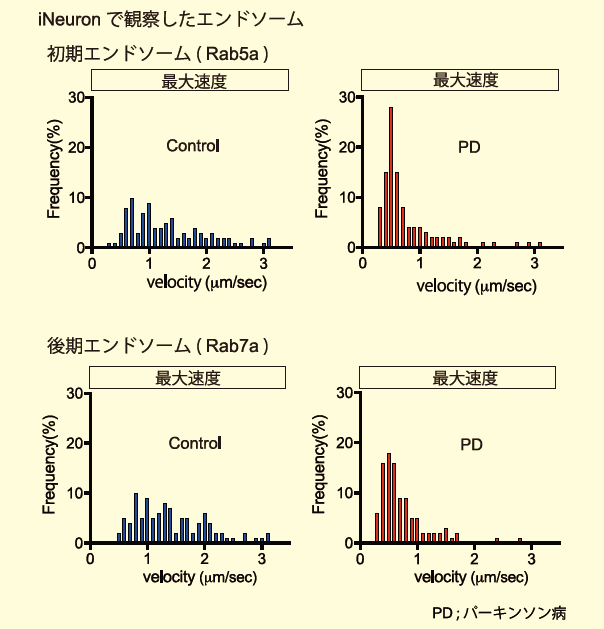
図3.
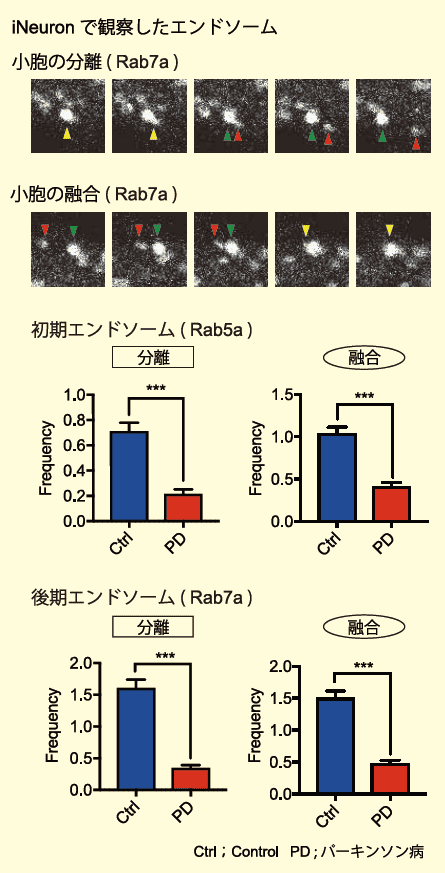
図4.
レトロマーの代表的な積荷タンパク質として知られるCI-MPR(cationindependent mannose 6-phosphate receptor)は、パーキンソン病の病因タンパク質であるα-シヌクレインの代謝酵素であるカテプシンDをリソゾームに輸送する受容体である。そこで、iPS細胞から分化誘導したグリア細胞におけるCI-MPRの細胞内局在を調べたところ、疾患群ではゴルジ体近傍に貯留している傾向があり、患者細胞ではCI-MPRの輸送障害が生じている可能性が示唆された。
パーキンソン病の病理学的特徴は神経細胞内のα-シヌクレインの異常蓄積である。そこで、iPS細胞由来ドパミンニューロンを免疫染色し、細胞質におけるα-シヌクレイン輝度値を測定したところ、疾患群において優位に高かった。この結果から、VPS35(D620N)変異陽性患者のドパミンニューロンではα-シヌクレインの細胞内蓄積が亢進している可能性が示された。
終わりに
iPS細胞技術は、従来の研究では困難だったヒト神経系細胞を用いたin vitro疾患モデルの解析を可能にし、病態解明に向け新たな研究手法の開発に道を開いている。今後、疾患特異的iPS細胞を利用することにより、病態発症メカニズムに即した治療薬開発が加速すると期待されている。しかし、データの再現性を担保するためには分化した細胞を長期間に渡って培養し安定した状態で維持する必要があり、培養期間は時として2ヶ月間に及ぶこともある。特にライブイメージングを行うにあたっては分化・成熟したiNeuronを長期間維持できる培養条件を確立することが必須であるが、近年開発された神経細胞用培地*により容易にニューロンを培養できるようになったことは研究者にとって朗報である。
* 本研究ではiNeuronの維持培養に神経細胞用培地(富士フイルム和光純薬コードNo. 148-09671)を使用した。研究の詳細については以下の論文を参照。
Bono, K. et al . : Mol. Brain, 13, 137 (2020).
参考文献
- Jankovic, J. : J. Neurol. Neurosurg. Psychiatry, 79, 368 (2008). DOI: 10.1136/jnnp.2007.131045
- Spillantini, M. G. et al. : Nature, 388, 839 (1997). DOI: 10.1038/42166
- Deng, H. et al. : Ageing Res. Rev., 42, 72 (2018). DOI: 10.1016/j.arr.2017.12.007
- Athauda, D. and Foltynie, T. : Nat. Rev. Neurol., 11 (1), 25 (2015). DOI: 10.1038/nrneurol.2014.226
- Okano, H. and Yamanaka, S. : Mol. Brain, 7, 22 (2014). DOI: 10.1186/1756-6606-7-22
- Vilariño-Güell, C. et al. : Am. J. Hum. Genet., 89, 162 (2011). DOI: 10.1016/j.ajhg.2011.06.001
- Zimprich, A. et al. : Am. J. Hum. Genet., 89, 168 (2011). DOI: 10.1016/j.ajhg.2011.06.008
- Ando, M. et al. : Mov. Disord., 27, 1413 (2012). DOI: 10.1002/mds.25145
- Seaman, M. N. et al. : J. Cell Biol., 142 (3), 665 (1998). DOI: 10.1083/jcb.142.3.665
- Hierro, A. et al. : Nature, 449, 1063 (2007). DOI: 10.1038/nature06216
- Burd, C. and Cullen, P. J. : Cold Spring Harb. Perspect. Biol., 6, a016774 (2014). doi: 10.1101/cshperspect.a016774
- Abeliovich, A. and Gitler, A. D. : Nature., 539, 207 (2016). DOI: 10.1038/nature20414
- Galvez, T. et al. : Cell, 151, 234 (2012). DOI: 10.1016/j.cell.2012.09.013