【連載】The Gateway to qNMR ~定量NMRへの扉~ 「第 2 話 qNMR で精確な測定をするために」
本記事は、和光純薬時報 Vol.85 No.4(2017年10月号)において、和光純薬工業株式会社 試薬化成品研究所 三浦 亨が執筆したものです。

qNMR の手法は、大きく分けて内標準法と外標準法の 2 種類があります。試料溶液に qNMR 用基準物質を内標準物質として添加して測定するのが内標準法です。これに対して外標準法は、試料溶液とは別に qNMR 用基準物質を一定量溶かした濃度既知の標準溶液を用意して、試料溶液と標準溶液を別々に測定し定量する手法です。外標準法は試料に qNMR 用基準物質を加える必要がなく、試料をそのまま回収できる点が大きな利点です。内標準法と外標準法の測定パラメータは基本的に同じと考えることができますが、外標準法によって純度や含有率を測定する場合、計算式によって補正される項に加えて、NMR 試料管の違いなどを考慮する必要があるので、内標準法と比べて操作が複雑です。したがって、外標準法は内標準法と比べて、一般的に精度の保証が難しいといわれています。
そこで今回は、日本薬局方に採用済みの内標準法の測定フローに従って、qNMR で精確な測定をするためのポイントをご紹介したいと思います。内標準法の測定フロー概略図を Figure 1 に示しました。試料調製、NMR 測定、データ解析の 3 つのパートに分かれていますが、ご覧の通り、測定フローはいたって単純です。実際には、これらの操作で得られた天秤での秤量値や NMR スペクトルの信号面積などの値を Figure 2 の計算式に代入して分析結果を得ることになります。qNMR と聞くと NMR 測定だけを正確に行えば良いと思われがちですが、実際には試料調製、NMR測定、データ解析のすべてのパートを定量に適した方法で実施しなければ、最終的に精確な分析結果を得ることは困難です。ここからは、それぞれのパートに分けて精確な測定をするためのポイントをご紹介したいと思います。
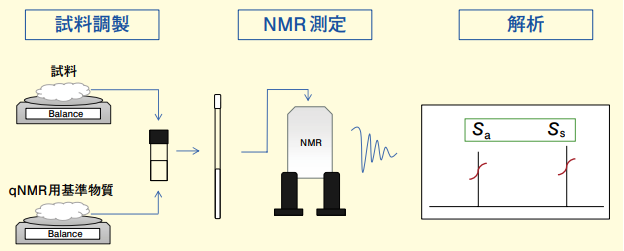
Figure 1.内標準法の測定フロー概略図
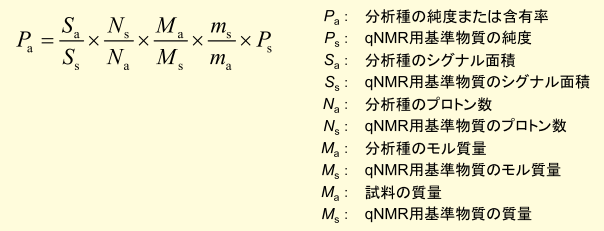
Figure 2.計算式
試料調製

Figure 3.日本薬局方(E)- ケイ皮酸 qNMR 純度規定の試料調製
出典:「第十七改正日本薬局方」(厚生労働省)(http://www.mhlw.go.jp/stf/seisakunitsuite/bunya/0000066530.html)qNMR の操作の中で軽視しがちなのが試料調製です。NMR を長年扱ってこられた方からすれば試料調製なんて"混ぜれば終わり"と思われるかもしれませんが、実は重要な操作です。日本薬局方(E)- ケイ皮酸 qNMR 純度規定の試料調製を Figure 3 に示しました 1)。この中では、ウルトラミクロ天秤(日本薬局方の記載では"ウルトラミクロ化学はかり"となっています)の使用が規定されていますが、これは、数 mg の試料や qNMR 用基準物質を精確に計量するためには、このぐらい高精度な天秤を使用しないと精確な分析結果を得ることが難しいという理由からです。
なお天秤での採取量の目安の参考になるものとして、米国薬局方(United States Pharmacopeia;USP)の最小計量値(minimum weight;Wmin)があります。これは、天秤での計量の要求精度を満たす下限値のことで、はかりとる質量は、最小計量値より大きいことが目安となります。最小計量値 Wmin は、下記 Eq.(1)に示したように、風袋など(一例として、試料採取用容器として用いるアルミ秤量皿(約 60 mg)などがあげられます)の質量 を 10 回繰り返し測定した際の標準偏差(standard deviation;SD)σに、安全係数 2 を計量における許容範囲(0.10 %)で除した値 2000 (= 2/(0.10/100))を乗じて算出することができます 2)。みなさんも普段使っている天秤で最小計量値を一度測定してみることをお勧めします。
Wmin=σ× 2000 Eq. (1)
また、使用する qNMR 用基準物質についても注意が必要です。昨今、qNMR 用基準物質として複数の製品が供給されていますが、その中でも国際単位系(International System of Units;SI)にトレーサビリティ(この場合は物質量(モル;mol))が確保された認証標準物質(Certified Reference Material;CRM)を使用することが望ましいと言えます。qNMR の測定原理は、NMR スペクトル上に観測された物質量が既知の基準物質の信号面積と分析種の信号面積を比較することで、分析種の物質量をもとめる方法ですので、物質量の基準となる qNMR 用基準物質としては、やはり SI トレーサブルな純度が精確に値付けされている認証標準物質の使用をお勧めします。
qNMR 用基準物質に求められるその他のポイントとしては、高純度なもの、安定して精確に秤量できるもの、信号の数やスピン結合による信号の分裂が少ないもの、特異的な領域に化学シフトを与えるもの、重溶媒中で安定かつ試料と反応しないもの、重溶媒への溶解性が良好であるもの、などが挙げられます 3)。
試料調製におけるその他のポイントとしては、使用する重水素化溶媒由来の信号が、定量分析の対象となる信号に重ならないことや、試料と qNMR 用基準物質が重水素化溶媒に完全に溶解していることなどが挙げられます。前者に関しては、ブランクとして重水素化溶媒だけの NMR スペクトルを取得し確認することをお勧めします。後者に関しては、溶媒の種類、温度、pH などさまざまな条件で溶解性が異なるため、溶解性に関する情報があれば参考にすることができます。
NMR 測定

Figure 4.日本薬局方(E)- ケイ皮酸 qNMR 純度規定の試験条件
出典:「第十七改正日本薬局方」(厚生労働省)(http://www.mhlw.go.jp/stf/seisakunitsuite/bunya/0000066530.html)qNMR に用いる NMR 測定条件としては、日本薬局方 qNMR 純度規定の試験条件が参考になります。Figure 4 に示した日本薬局方(E)- ケイ皮酸 qNMR 純度規定の試験条件を参考に、NMR 測定条件についてポイントをご紹介したいと思います 1, 4)。
①装置は 1H 共鳴周波数 400MHz 以上と設定されています。NMR の感度と分解能は外部磁場が大きくなるにしたがって向上するため、十分な感度と分解能が得られるように 400MHz 以上に設定されています。装置の共鳴周波数は、測定の目的(目標とする精度など)に合ったものを選択することをお勧めします。
②デジタル分解能は 0.25Hz 以下に設定されています。NMR 信号は最初アナログ信号として取得されますが、その後デジタル信号に変換されます(パルス FT-NMR を前提としています)。その際、データポイント数が少ないと、フーリエ変換後の NMR スペクトルにおける信号の形状を正しく再現することができません。日本薬局方では信号の形状を再現するための十分なデジタル分解能として 0.25Hz 以下が設定されています。
③スピニングをオフ(試料管の無回転)に設定しているのは、試料管を回転したときに現れるスピニングサイドバンドが、定量分析の対象となる信号に重なる可能性があり、これを回避するのが目的です。
④パルス角が 90°に設定されているのは、単位時間当たりの感度が良く、積算効率が高いからです。
⑤ 13C 核デカップリングはありに設定されています。13C 核デカップリングとは、13C 核とのスピン結合によって起こる微小な信号の分裂(13C サテライトサイドバンド)を解消する手法です。13C サテライトサイドバンドが、定量分析の対象となる信号に重なることで精確な信号面積が得られなくなるのを防ぐことが目的です。
⑥遅延時間は繰り返しパルス待ち時間 60 秒以上に設定されています。遅延時間とは、パルス系列において最初のパルスを打ってから、次の積算で同じパルスを打つまでの待ち時間です。定性測定などで一般的に用いられる遅延時間は 5 秒程度と短いですが、qNMR では信号面積の飽和を防ぐために、有効数字 2 桁以上の定量精度を目標とするとき、遅延時間を縦緩和時間(spinlattice relaxation time、T1)の 7 倍以上設定する必要があります 5)。日本薬局方では、通常の化合物の T1 が長いものでも 7 秒程度ですので、その 7 倍を十分に確保している 60 秒以上を設定しています。
データ解析
データ解析における主なポイントを以下に記載します。まず窓関数(ウィンドウ関数)処理について注意が必要です。窓関数とは、FID 信号をフーリエ変換するときに、スペクトルの感度や線幅などを改善させるために掛け合わせる関数のことを言います。窓関数処理をすることによって目的に合ったスペクトルに加工することができますが、その一方で、不適切な窓関数処理によって信号面積の精確さを失うことがあります。qNMR では、精確さを検証している場合を除いて、窓関数処理を行わないことが望ましいと言えます。
ゼロフィリング処理は、データポイントが補完され、スペクトル形状が滑らかになることで、信号面積の精度が向上するので、実施することをお勧めします。なおゼロフィリングとは、得られた FID 信号の後ろに強度ゼロのデータポイントを補完し、見かけ上のデジタル分解能を向上させる方法のことです。
また NMR 信号の位相は NMR 信号取得前には不明であるため、測定後に位相を合わせる必要があり、この操作を位相補正といいます。解析ソフトウェアによって、NMR 信号の位相補正を自動的に行うことが可能ですが、位相のずれが残ることが多々あります。qNMR では、位相のずれは信号面積のばらつきの原因となるため、手動操作で位相のずれを解消する必要があります 6)。
NMR スペクトルのベースラインは必ずしも強度 0 とならない場合が多いです。これは信号面積の取得においてばらつきの要因となるため、qNMR ではベースライン補正を行うことをお勧めします 6)。ベースライン補正には様々なアルゴリズムがありますが、qNMR では、ベースラインと見なされるデータポイントを補正点として利用してベースラインを平らにする処理を行うことをお勧めします。
選択したアルゴリズムによっては、信号の裾の一部などのデータポイントが補正点として利用され、その結果、信号形状が大きく影響を受けて信号面積が極端に変わる場合があるので注意が必要です。ベースライン補正は、信号面積に大きな影響を与える場合があるため、一定のルールを決めて慎重に行うことが望ましいといえます。
次回は、実際の測定例を中心にお話ししたいと思います。
参考文献
- The Japanese Pharmacopoeia, Seventeenth Edition (E)-Cinnamic acid for assay 2 (Purity value by quantitative NMR).
- USP "General Chapter 41 Balances", US Pharmacopeia USP 39-NF 34(2016).
- Miura, T., Sugimoto, N., Suematsu, T., Millis, K. K., Asakura, K. and Yamada, Y. : New Horizons of Process Chemistry, 275- 285 (2017).
- Hosoe, J., Sugimoto, N., Suematsu, T., Yamada, Y., Miura, T., Hayakawa, M., Suzuki, H., Katsuhara, T., Nishimura, H., Kikuchi, Y., Yamashita, T. and Goda, Y. : Pharmaceutical and Medical Device Regulatory Science, 45 , 243- 250 (2014).
- Saito, T., Nakaie, S., Kinoshita, M., Ihara, T., Kinugasa, S., Nomura, A. and Maeda, T. : Metrologia, 41 , 213- 218 (2004).
- Yamazaki, T., Saito, T., Miura, T. and Ihara, T. : BUNSEKI KAGAKU, 61 , 963- 967 (2012).



