【総説】環状ペプチド合成に有用なジスルフィド形成試薬 Npys-OMe の創製
本記事は、和光純薬時報 Vol.86 No.4(2018年10月号)において、東京薬科大学 薬学部 薬品化学教室 田口 晃弘様、林 良雄様、東京薬科大学 薬学部 分析化学教室 小谷 明様、袴田 秀樹様に執筆いただいたものです。
はじめに
ペプチドやタンパク質に見られるジスルフィド結合は、ペプチド鎖の環状化により立体構造を固定化することで、その生理機能の発現に寄与する重要な官能基である。当該結合を有する生理活性ペプチドは数多く知られ、インスリン、カルシトニンやナトリウム利尿ペプチドなどは医薬品として臨床適用されている。
このような環状ペプチドの化学合成では、ジスルフィド架橋は非常に重要な工程となる。そのためペプチド化学は、これまで多くのジスルフィド架橋法を開発してきた。一般に 1 組のジスルフィド結合の構築には、従来法として空気酸化(DMSO酸化)やヨウ素(I2)酸化が多用されるが、複数のジスルフィドを有するペプチドの場合、当該結合を一挙に構築すると架橋異性体が副生しやすい。そこで、位置選択的なジスルフィド架橋法として、システイン(Cys)残基側鎖チオール(SH)基の直交型保護基を利用した段階的ジスルフィド架橋法が開発されてきた。
1980 年に松枝らによって開発された 3- ニトロ -2- ピリジンスルフェニル(Npys)基 1)は、ペプチド合成において Cys 残基側鎖 SH 基の直交型保護基の一つとして利用されるが、同時に活性ジスルフィドとしても機能する 2)。そのため、他に側鎖無保護のCys 残基が共存すれば、ジスルフィド交換反応が選択的に進行し、ペプチド内あるいはペプチド間でジスルフィド結合が形成される(図 1A)。
我々は、この Npys 基の化学に着目し、ケミカルバイオロジー研究やペプチド合成化学に有用な、固相担持型ジスルフィド形成試薬の開発を精力的に行ってきた 3-5)。その開発の中で、硫黄−酸素結合を有する 3- ニトロ -2- ピリジンスルフェン酸エステル(Npys-OR)の構造に興味を持った(図 1B)。
当該エステルの合成法は松枝らにより報告 6)されているが、その性質に関しては研究がなされていなかった。
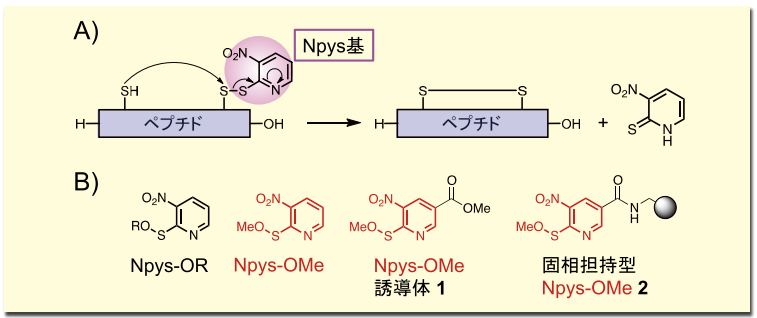
図1.
A)3- ニトロ -2- ピリジンスルフェニル(Npys)基によるジスルフィド結合の構築。B)3- ニトロ -2- ピリジンスルフェン酸メチルエステル(Npys-OMe)およびその誘導体 1, 2 の化学構造。
今回我々は、Npys-OR のジスルフィド形成における酸化剤としての可能性を模索した。その結果、3- ニトロ -2-ピリジンスルフェン酸メチルエステル(Npys-OMe)およびその誘導体 1, 2(図 1B)が、環状ジスルフィドペプチドの合成に有用なジスルフィド形成試薬となることを新たに見出した 7, 8)。本稿では、これらの試薬の合成、および環状ジスルフィドペプチド合成への応用について紹介する。
Npys-OMe誘導体の合成7)
Npys-OMe 誘導体 1 および固相担持型 Npys-OMe 2 の合成を Scheme 1に示す。誘導体 1 の合成では、まず、6- ベンジルチオ -5- ニトロニコチン酸メチル(3)のベンジル基を 1,2- ジクロロエタン(1,2-DCE)溶媒中、ピリジン存在下、塩化スルフリル(SO2Cl2)と反応させ、クロロ体 4 へと導いた。
得られた 4 を精製すること無く、THF 溶媒中、メタノールおよび塩基で処理することで誘導体 1 を合成した(2 工程 79%)。一方で、固相担体として水系および有機溶媒にて使用可能なChemMatrix® resin を用い、固相担持型 Npys-OMe 2 を合成した。樹脂 55)よりクロロ化、続くメタノール処理により樹脂 2 を得た(Scheme 1B)。
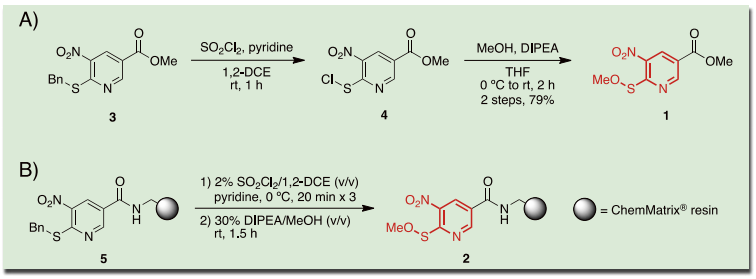
Scheme 1.
A)Npys-OMe 誘導体 1 および、B)固相担持型 Npys-OMe 2 の合成 7)Npys-OMe 誘導体 1 のジスルフィド形成能評価 7)
還元型オキシトシン(6)からオキシトシン(7)への酸化反応をモデルとし、Npys-OMe 誘導体 1 のジスルフィド形成能を評価した。反応中に生成するオキシトシンを HPLC にて追跡し、検量線に基づき HPLC 収率を算出した。表 1 にその酸化反応の結果を示す。
表1.オキシトシン合成によるジスルフィド形成能の評価 7)
[a] 反応 6 時間後におけるオキシトシン(7)の HPLC 収率。
[b] オリゴマーの HPLC 収率。
[c] 反応 3 時間後におけるオキシトシン(7)の HPLC 収率。
| Entry | 1 (equiv.) |
Solvent | Peptide 6 conc. (mM) |
Yield of oxytocin (%)a |
Oligomer (%)b |
|---|---|---|---|---|---|
| 1 | 2 | DMF : H2O (1 : 1) | 1 | 77 | 16 |
| 2 | 2 | DMF : H2O (1 : 2) | 1 | 79 | 16 |
| 3 | 2 | CH3CN : H2O (1 : 2) | 1 | 86 | 14 |
| 4 | 2 | CH3CN : H2O (1 : 3) | 1 | 88 | 19 |
| 5 | 2 | CH3CN : H2O (1 : 3) | 0.5 | 87 | 8 |
| 6 | 2 | CH3CN : H2O (1 : 3) | 0.1 | 90 | 2 |
| 7 | 5 | CH3CN : H2O (1 : 3) | 0.1 | 92c | 1 |
| 8 | 0 | CH3CN : H2O (1 : 3) | 0.1 | 1 | 0 |
始めにペプチド 6 の DMF : H2O(1 : 1、ペプチド濃度 1 mM)溶液に対し、誘導体 1 を 2 当量添加し、室温にて 6 時間攪拌したところ、オキシトシンを収率 77% で得た(Entry 1)。また、Entry 2 および 3 に示すように反応溶媒を DMF : H2OからCH3CN : H2O に変更すると収率の向上が見られた。更にEntry 4の CH3CN : H2O(1 : 3)の条件下では、収率 88% と良好な結果を示した。
しかし、副生成物としてペプチド 6 のオリゴマーが観察されたため、これらの生成を抑制すべく、反応条件の最適化を図った。CH3CN : H2O(1 : 3)におけるペプチド 6 の濃度を 0.5 および 0.1 mM に調整し、酸化反応を実施したところ、ペプチド濃度の低減によりオリゴマーの生成は減少し、0.1 mM では 2% にまで抑えることができた。
またこれに伴い、反応 6 時間後のオキシトシン(7)の収率はそれぞれ 87、90% と高値であった(Entry 5 および 6)。更に、ペプチド 6 の CH3CN : H2O(1 : 3、ペプチド濃度 0.1 mM)溶液に対し、誘導体 1 を 5 当量用いると、3時間で反応は完結し、オキシトシンを 92% の収率で合成することができた(Entry 7, 図 2)。
一方で、Entry 8 の様に Npys-OMe 誘導体 1 を添加しないとオキシトシンがほとんど生成しないことから、pH 6.0 ではオキシトシンの空気酸化によるジスルフィド形成は起こらず、当該誘導体の添加によりはじめて酸化反応が効率的に進行することが示唆された。また、構造を簡素化させた Npys-OMe も、同様の反応条件にてオキシトシンを高収率(95%)で合成することが可能である(図 3)8)。
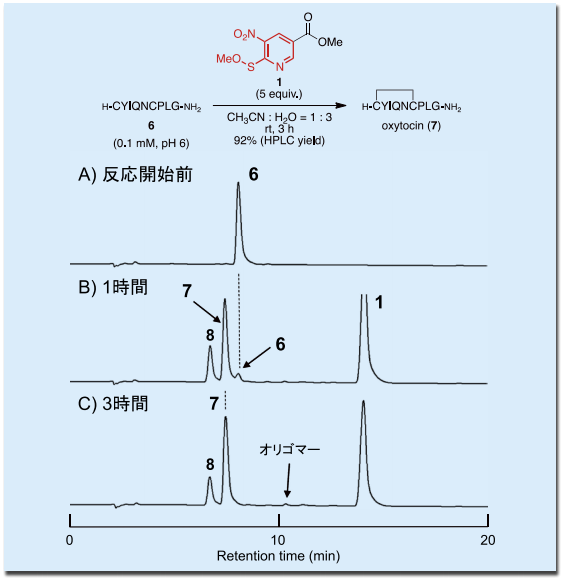
図2.
表 1、Entry 7の反応条件におけるオキシトシン合成のHPLC チャート 7)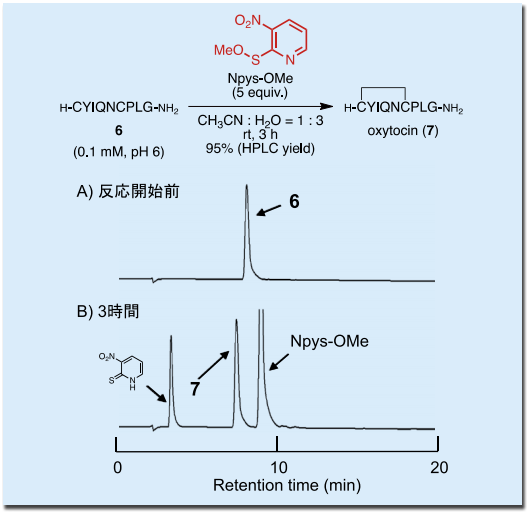
図3.
Npys-OMe によるオキシトシン合成 8)一方で、樹脂 2 を用いる条件でも良好な収率(85%)でオキシトシン(7)が生成する(Data not shown)。この方法では、試薬成分が樹脂上に残るため、反応溶液のろ過操作のみで、純度の高いペプチドの回収が可能である 7, 8)。
Npys-OMe 誘導体 1 の酸化還元特性の評価 7)
誘導体 1 のサイクリックボルタンメトリー 9)で観察された還元波のピーク電位(Epc)は−0.541 V であり、酸化型グルタチオン(GSSG)の−1.55 Vよりも正側であった(表 2)。この結果は、Npys-OMe 誘導体 1 により2つの SH 基をジスルフィドへと酸化可能であることを示唆している。
表2.
サイクリックボルタンメトリーで測定した還元波のピーク電位(Epc)7)
| 測定対象物質 | Epc(V vs. Ag/AgCl) |
|---|---|
| I2 | +0.437 |
| NCS | -0.175 |
| Npys-OMe 誘導体 1 | -0.541 |
| 酸化型グルタチオン(GSSG) | -1.55 |
ボルタンメトリー条件:作用電極,プラスチックフォームドカーボン;参照電極,Ag/AgCl;対極,白金;電解質溶液,0.1 M KCl 含有 20 mM リン酸緩衝液(pH 6.0):CH3CN(3:1, v/v);掃引速度,20 mV/s.
一方で、ジスルフィド形成に使われる既存の酸化剤 I2 および最近ジスルフィド形成試薬として報告されたN- クロロスクシンイミド(NCS)10)のEpc は、それぞれ +0.437 および−0.175 V であった。Npys-OMe誘導体 1 のEpc(−0.541 V)は、I2 および NCS の値よりもはるかに負側であることから、誘導体 1 は温和な酸化剤として機能することが電気化学的手法により確認された。
Npys-OMe 誘導体 1 を用いたペプチド合成 7)
我々は、Npys-OMe のペプチド合成化学への更なる応用に向け、オキシトシン(7)よりも複雑な構造を有する環状ペプチドの合成を試みた。
(1)ヒト心房性ナトリウム利尿ペプチド(hANP(9))の合成
還元型 hANP(10)の CH3CN : H2O(1 : 3、ペプチド濃度 1 mM)溶液にNpys-OMe 誘導体 1 を 5 当量添加し、室温にて撹拌した。図 4 に示すように時間経過に伴い、還元型ペプチド 10のピークは減少し、hANP(9)のピークの増大を確認できた。反応 24 時間後では、10 のピークは完全に消失した。また、本反応中、酸化されやすいアミノ酸の1つである 12 残基目のメチオニンには全く影響を与えなかった。反応溶液を HPLC にて精製することで 9 を単離収率 50% で得た。
本結果より、Npys-OMe 誘導体 1 は、中分子ペプチドのジスルフィド結合構築においても効率的に機能することが示された。
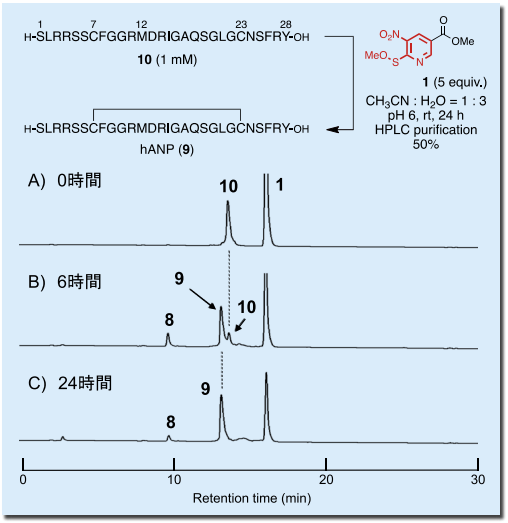
図4.
hANP(9)の合成 7)(2)α- コノトキシン ImI(11)の合成
続いて、2 組のジスルフィド結合を有するα- コノトキシン ImI(11)の合成検討を実施した(図 5)。保護Cys 残基を有する還元型ペプチド 12を用い、酸化反応を段階的に行うことで、位置選択的なジスルフィド架橋を試みた。即ち、まず、Npys-OMe 誘導体 1 により無保護 Cys2-Cys8 残基間を架橋した後、次いで I2 酸化により保護 Cys3-Cys12 残基間を架橋し、αコノトキシン ImI(11)の合成を目指した。
まず、Cys 保護ペプチド 12 の CH3CN : H2O(1 : 3、ペプチド濃度 1 mM)溶液に誘導体 1(2 当量)を添加し、室温にて撹拌した。時間経過に伴い、Cys2-Cys8 間でジスルフィド結合したペプチド 13 の生成が確認され、27 時間後でペプチド 12 のピークは消失した。また、本反応中では、オリゴマーや保護 Cys 残基間の架橋体などの副生成物は生じなかった。
HPLC 精製により、所望のジスルフィドペプチド13 を単離収率61% で得た。この結果から、誘導体1 は無保護SH基間の選択的なジスルフィド架橋形成に有効なことが、再び示された。
次に、ペプチド13 に対してI2 酸化を行い、アセトアミドメチル(Acm)基で保護されたCys 残基間を架橋した11 を単離収率50% で得た。本方法にて得られたペプチド11 と標品とのHPLC の重ね打ちにより、ピークが一致したことから、構築した2 組のジスルフィド結合は位置選択的であることが示された。この結果から、Npys-OMe 誘導体による酸化法と他の酸化法を組み合わせることで、ジスルフィド結合の新たな位置選択的合成法の提供が可能となった。
おわりに
今回、Npys-OMe およびその誘導体が、温和なジスルフィド形成試薬として機能することを見出した。本試薬を用いると、比較的高濃度な還元型ペプチド溶液においても、環状ジスルフィドペプチドを効率的かつ簡便に得ることが可能である。従って、有機化学合成または創薬研究を行う研究者が用いる試験研究用試薬として、また製薬企業での医薬品等の製造における反応試薬として、本試薬は高い有用性が期待できる。
また、Npys-OMe は安定な化合物で、取り扱いも容易であることから、ジスルフィド化合物を扱われる研究者の方に幅広くご利用いただければ幸いである。
キーワード
サイクリックボルタンメトリー
静止溶液中に電極を配置し、電極電位を繰り返し掃引しながら電流を測定する電気化学分析法である。本法は、電気化学的に活性な化学種の電極反応を把握するために最初に行う方法であり、酸化還元電位や電極反応の可逆性などの情報も得られるので、様々な研究領域において多用されている。
参考文献
- Matsueda, R. et al.: Int. J. Pept. Protein Res., 16, 392 (1980). DOI: 10.1111/j.1399-3011.1980.tb02963.x
- Bernatowicz, M. S. et al.: Int. J. Pept. Protein Res., 28, 107 (1986). DOI: 10.1111/j.1399-3011.1986.tb03236.x
- Fukumoto, K. et al.: Tetrahedron Lett., 53, 535 (2012). DOI: 10.1016/j.tetlet.2011.11.089
- Fukumoto, K. et al.: Asian J. Org. Chem., 4, 1030 (2015). DOI: 10.1002/ajoc.201500267
- Taguchi, A. et al.: Org. Biomol. Chem., 13, 3186 (2015). DOI: 10.1039/C5OB00030K
- Matsueda, R. et al.: Heterocyclces, 15, 1089 (1981). DOI: 10.3987/S-1981-02-1089
- Taguchi. A. et al.: Chem. Eur. J., 23, 8262 (2017). DOI: 10.1002/chem.201700952
- 林 良雄、田口 晃弘、福元 謙太郎:特願 2016-101812, PCT/JP2017/019086.
- Kotani, A. et al.: Electrochemistry, 83, 363 (2015). DOI: 10.5796/electrochemistry.83.363
- Postma, T. M. et al.: Org, Lett., 15, 616 (2013). DOI: 10.1021/ol303428d
