【総説】ラセミ体アルコールを光学的に純粋な化合物に収率 100%で 変換する新技術
本記事は、和光純薬時報 Vol.85 No.1(2017年1月号)において、大阪大学大学院薬学研究科 赤井 周司 様に執筆いただいたものです。
はじめに
光学的に純粋な有機化合物は、医薬品、農薬、香料、液晶などの様々な製品に用いられている。年々、光学活性化合物の需要は高まり、環境に負荷をかけない供給法の開発が今日の重要課題の1つである。最近の金属触媒や低分子有機触媒の発展には目を見張るものがあるが、常温常圧での高いエナンチオ並びに官能基選択性、高い触媒回転率を満足するものは未だ少ない。一方、生体触媒(酵素)はこれらを可能にする。さらに、発酵によって大量生産でき、かつ、生分解されて自然に帰すことができる酵素は、枯渇しない触媒として資源の乏しいわが国の今後の物質生産・変換を支える鍵になる。
多様な酵素の中でも、脂質のエステル結合を加水分解するリパーゼは、有機合成への利用が最も多い。それは、リパーゼは特に安定で、補酵素が要らず、かつ、非天然の幅広い基質に対して高い触媒活性とエナンチオ選択性を示すからである。さらに、リパーゼを有機溶媒中で用いると、カルボン酸とアルコールからエステルを形成する触媒にもなる。
その際、d 体と l 体の2つの鏡像異性体の片方だけを選択的に反応させることができるエナンチオ選択性のために、ラセミ体を分離し光学活性体を入手する速度論的光学分割に汎用されてきた (図1A)。
最近では、何十種類ものリパーゼが市販されており、セライトや樹脂などに担持され粉末状になっているものが多い。このように、リパーゼは極めて使い勝手がよい生体触媒で、工業生産への適用例も多いが 1)、改良の余地も有る。
その1つは収率である。リパーゼはラセミ体の片方の鏡像異性体だけを反応させるために、生成物の収率は最大 50% になる。反応しなかった残りの半分を再利用できれば収率 100% を達成できるが、それを1つのフラスコ内で行うことができれば、より実用的な変換法になる。
本稿では、著者らが最近開発した『ラセミ体アルコールを光学的に純粋な化合物に収率 100%で変換する動的光学分割法』について紹介する。
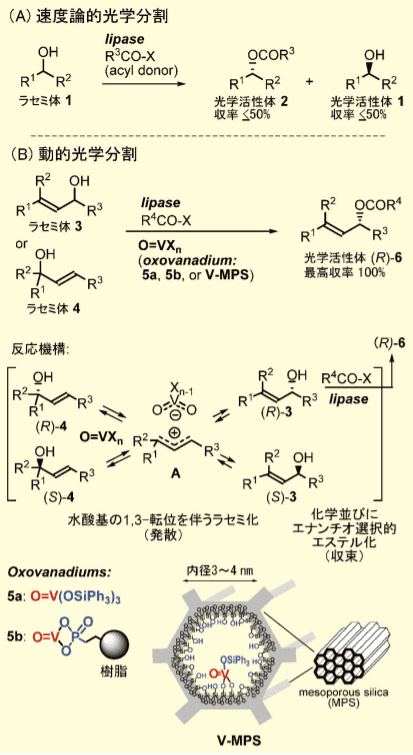
図1.
(A)リパーゼ触媒速度論的光学分割(B)オキソバナジウムとリパーゼを併用する動的光学分割
リパーゼ触媒による動的光学分割(DKR)法の背景 2)
リパーゼ触媒による速度論的光学分割と並行して、反応せずに残った鏡像異性体を別の触媒によってラセミ体に戻すこと(ラセミ化)が進行すれば、最終的には、すべてのラセミ体原料を1つの光学活性体に収率 100% で変換することができる。この手法は動的光学分割(dynamic kinetic resolution、以下 DKR と略す)と呼ばれる。
DKRを成功させるためには幾つかの要件が同時に満たされなければならない(詳細は総説 2)参照)が、なかでも最も難題は、酵素とラセミ化触媒が1つのフラスコ内で共存し、本来の機能を発揮できることである。ラセミ化効率を高めるために触媒の活性を上げると、リパーゼとの共存性が低下する。その理由は、ペプチドであるリパーゼがもつ官能基によって両触媒は反応して失活したり、リパーゼが触媒活性を維持するために表面に抱えている多数の水分子によってラセミ化触媒が失活することがあるためである。
現在の DKR の主流は、ルテニウム錯体とリパーゼを併用する手法である。ルテニウム錯体はアルコールの酸化-還元反応によってラセミ化を触媒する。ルテニウム錯体とリパーゼは共存性に概ね優れ、本法の適用範囲は広い 2)。
著者らの DKR 法と有機合成化学的応用
著者らは最近、前述のルテニウム錯体を用いる方法とは全く異なる DKR 法を開発した。以前より、オキソバナジウムエステル O=V(OR)3 はアリルアルコールと反応して、水酸基の 1,3-転位を起こすことが知られていたが、150℃以上の高温を要した 3)。
著者らは、この転位反応を再検討した結果(図1B)、O=V(OSiPh3)3 5a4)又は樹脂に結合したホスホン酸のオキソバナジウムエステル 5b5) を用いると、アセトン、アセトニトリルなどの極性溶媒中、室温〜 50℃でアリルアルコール(3 及び 4)の 1,3- 転位反応が進行することを見出した。
しかも、光学活性アルコールは転位の間にラセミ化することを新たに発見した。種々の実験結果から、イオン対中間体 A が生じ、その後の再結合の際にラセミ化が起こっていることがわかった。このラセミ化とリパーゼ触媒光学分割を組み合わせることで DKR が達成できた。すなわち、ラセミ体の 3 に、5a 又は 5b(10 mol%)、市販の固定化リパーゼ、及びアシル化剤として酢酸ビニルを反応させると、DKR が進行し光学純度 90 〜 99% ee のエステル(R)-6 が収率 64 〜 99%で得られた。
これは、オキソバナジウムが、ラセミ化を伴いながら 4 種の異性体 [(R)-3,(S)-3,(R)-4,(S)-4] 間の動的平衡を生じ(発散)、リパーゼがその混合物のなかから(R)-3 を高選択的に変換する(収束)という相反する性質の反応が同時進行することで初めて達成された(図1B)。各触媒単独ではこの成果は得られない。
また、本法では 3 の位置異性体 4 が等価な原料として利用できることが合成化学上の大きな利点である(合成化学的応用例は後述)。しかし、オキソバナジウム (5a, 5b)のラセミ化能は必ずしも充分でなく、基質によってはラセミ化が遅いために生成物の収率や光学純度が低いことが問題であった。一方で、よりラセミ化能の高いオキソバナジウムを用いると、リパーゼとも反応して互いを失活させた。
ラセミ化活性とリパーゼとの共存性を同時に向上させたラセミ化触媒を開発すべく、種々検討した結果、メソポーラスシリカ(MPS)の細孔を利用して両触媒を物理的に隔離する案に至った。MPS は二酸化ケイ素(シリカ)を材質として、均一で規則的な細孔をもつ多孔質無機化合物で、表面積が約 1000 m2/g もある。
また、細孔径が 2 〜 50 nm の異なる種々の MPSを入手することができるが、本 DKRでは細孔径 3 〜 4 nm の MPS を利用した。MPS と 5a を有機溶媒中で 80 ℃に加熱することで、細孔内表面のシラノールにバナジウムが共有結合した V-MPS(バナジウム含量 0.20 〜 0.22 mmol/g)を調製した。
その構造は ICP-AMS、元素分析、BET 法などによって図 1B のように推定している。分子量数百以下の有機化合物は V-MPS の細孔に簡単に出入りすることができるが、分子量数万ダルトン以上の巨大なリパーゼは細孔に入っていけない (実際には、ポリマーや無機素材に担持された市販のリパーゼを利用するので、細孔内に入る可能性は限りなくゼロに近い)。こうして、バナジウムによる『ラセミ化の反応場』と、リパーゼによる『光学分割の反応場』が MPS の細孔によって完全に分離できる (図 2)。
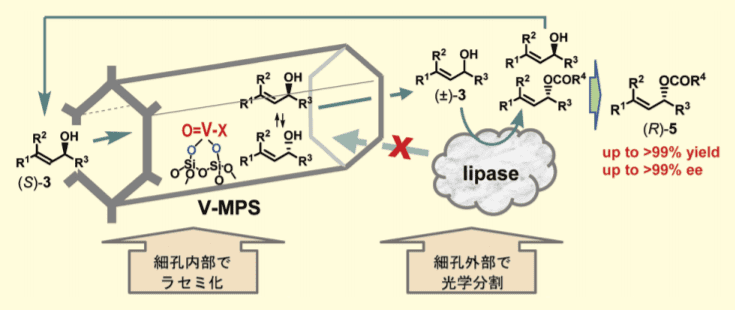
図2.MPS の細孔を利用する反応場の分離
新規に調製した V-MPS は、従来使用していたラセミ化触媒(5a, 5b)よりも格段にラセミ化活性が高いことがわかった 6a)。さらに、100 nm や 400 nm の大きな細孔径を有するマクロポーラスシリカに結合したオキソバナジウムはラセミ化活性が低いことから、V-MPS のナノサイズの細孔がラセミ化の促進に極めて重要な役割を果たしていることが明らかになった 6a)。
V-MPS と市販の固定化リパーゼは何れも粉末状である。これらを使ってラセミ体 4a の DKR を行った後、溶液部分と沈殿物を分離した。溶液を減圧濃縮すると、高純度の 6a が単離収率 99%、光学純度 99% ee で得られた(図 3A)。
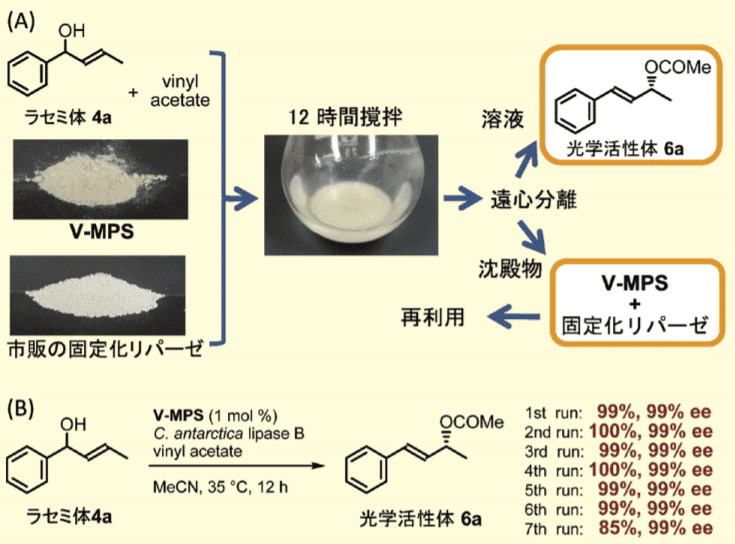
図3.
(A)V-MPS と市販の固定化リパーゼを用いる DKR の概要(B)触媒の回収再利用の具体例
また、溶液部分に漏洩したバナジウム量は 0.0003% 未満であった。 さらに、沈殿物(V-MPS とリパーゼの混合物)を減圧乾燥後、同じ条件下、再度 DKR に使用すると全く同じ結果が得られた。この、混合触媒は 6 回目まで全く失活することなく再利用できた(図3B)6a)。
さらに、細孔径が各々 3 nm 及び 4 nm の MPS から調製した V-MPS3 と V-MPS4 を用いて、分子サイズの異なる光学活性アルコールのラセミ化速度を比較した結果、分子量 200 程度の小さなアルコールでは、V-MPS3 と VMPS4 のラセミ化活性に差はなかったが、分子量が 400 を越え、また、分子長が 1 nm を越えるようなアルコールになると、V-MPS4 の方がラセミ化が速いことがわかった 6b)。
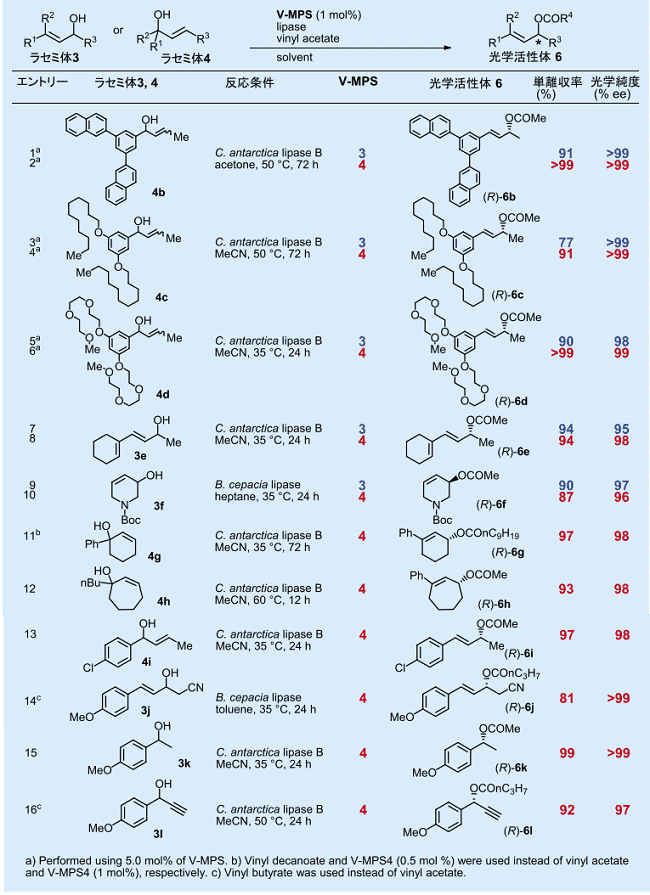
基質アルコールに適した市販の固定化リパーゼを選択し、これと V-MPS3 または V-MPS4 を併用する DKR の実施例の一部を表1に示す 6b)。前述のラセミ化速度の違いを反映して、4b 〜 4d のように比較的大きなアルコール分子では、V-MPS4 を用いると、より高収率で対応するエステルが得られる (エントリー 1 〜 6)。
一方、比較的小さなアルコールでは、V-MPS3 と V-MPS4 に殆ど差はない(エントリー 7 〜 10)。 従って、一般的には、V-MPS4 を用いることが推奨される。また、本法はベンジルアルコール 3k やプロパルギルアルコール 3l にも適用できる(エントリー 15, 16)。
表1.V-MPS と市販の固定化リパーゼを用いる DKR の実施例
本 DKR 法の応用例を 2 つ挙げる (図4)。(-)- クリナンの不斉合成では、シクロヘキセノンから 1 工程で得られるラセミ体第三級アルコール 4m に本法を適用し、アリルエステル(R)-6m(光学純度 98%)を得た。続いて、Ireland-Claisen 転位を行ってカルボン酸(S)-7 に誘導した後、4 工程の変換を行い、全合成を達成した(図 4A)7)。また、(R)- インペラネンの不斉合成では、バニリンから誘導したラセミ体アリルアルコール 3n に本法を適用し、アリルエステル(S)-6n(光学純度>99%)を得た。これをエポキシド(S)-8 に変換後、位置および立体選択的に Grignard 試薬を付加し、脱保護を経て全合成を達成した(図4B)6a)。
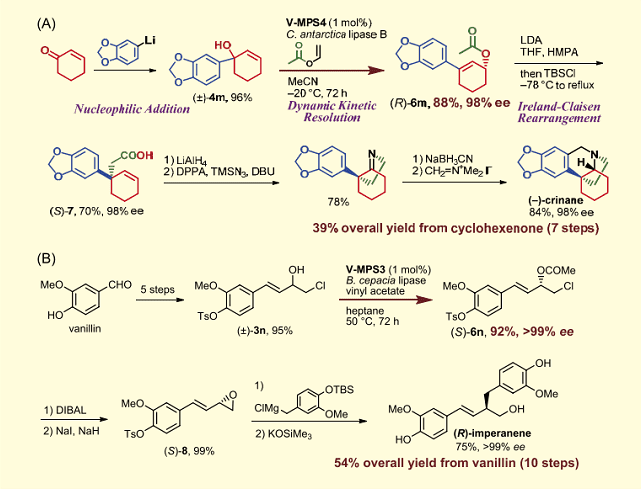
図4.V-MPS と固定化リパーゼを併用する DKR の応用
(A)(-)- クリナンの不斉全合成(B)(R)- インペラネンの不斉全合成
これらの事例は、入手容易なラセミ体アルコールが DKR によって、光学的に純粋な化合物に高収率で変換できることを示している。さらに前者は、DKR によって導入されたアシル基を分子骨格構築に有効活用した例であり(著者らは、類似の利用例を他にも報告している 8))、エノン、炭素陰イオン、アシル化剤の 3 成分をカルボニル炭素上でエナンチオ選択性に連結して全炭素置換光学活性第四級炭素を不斉構築する新手法として、今後の展開が期待できる。
まとめ
リパーゼ(生体触媒)とオキソバナジウム化合物(金属触媒)という異質な触媒を1つのフラスコ内で同時に用いることにより、入手容易なラセミ体アルコールを光学的に純粋な化合物にほぼ定量的に変換する手法を開発した。これは、オキソバナジウムが、ラセミ化を伴いながら複数の構造異性体間の動的平衡を生じ(発散)、リパーゼがその混合物のなかから1つの鏡像異性体を高選択的に変換する(収束)という相反する性質の反応が同時進行することで初めて達成された(図1B)。
また、高活性な 2 種類の触媒が互いに悪影響を及ぼすことなく共存し、本来の機能を最大限に発揮するためには、メソポーラスシリカのナノスケールの細孔を利用することが極めて効果的であることもわかった。著者らは、このナノスケールの反応場の更なる活用研究を進めている。
参考文献
- (総説)廣瀬芳彦:有合化誌 , 69, 506 (2011).
- (総説)(a) Akai, S. : Chem. Lett., 43, 746 (2014). DOI: 10.1246/cl.140223 (b) Takizawa, S. et al. : Chem. Eur. J., 21, 8992 (2015). DOI: 10.1002/chem.201406444 (c) Verho, O. and Bäckvall, J.E. : J. Am. Chem. Soc., 137, 3996 (2015). DOI: 10.1021/jacs.5b01031 (d) Långvik, O. et al. : ChemCatChem, 7, 4004 (2015). DOI: 10.1002/cctc.201500459
- Chabardes, P. et al. : J. Tetrahedron, 33, 1775 (1977). DOI: 10.1016/0040-4020(77)84059-3
- Akai, S. et al. : Angew. Chem. Int. Ed., 45, 2592 (2006). DOI: 10.1002/anie.200503765
- Akai, S. et al. : Org. Lett., 12, 4900 (2010). DOI: 10.1021/ol102053a
- (a) Egi, M. et al. : Angew. Chem. Int. Ed., 52, 3654 (2013). DOI: 10.1002/anie.201208988 (b) Sugiyama, K. et al. : Catal. Sci. Technol., 6, 5023 (2016). DOI: 10.1039/C6CY00257A
- Kawanishi, S. et al. : Green Chem., in press (2017).
- (a) Akai, S. et al. : Angew. Chem. Int. Ed., 43, 1407 (2004). DOI: 10.1002/anie.200353044 (b) Nemoto, H. et al . : Tetrahedron, 68, 7295 (2012). DOI: 10.1016/j.tet.2012.06.095