【連載】幹細胞由来EV~治療、診断、化粧品への展開~ 第4回 細胞外小胞の産業化に向けた標準開発の取り組み
本記事は、和光純薬時報 Vol.91 No.4(2023年10月号)において、富士フイルムホールディングス株式会社 知的財産部 国際標準化推進室 河内 幾生様に執筆いただいたものです。
はじめに
細胞は脂質二重膜を有する小胞にタンパク質や核酸などの機能分子を内包し、分泌している。分泌された小胞が、体液を通じて周囲の細胞や遠隔地の細胞へと運ばれ、その小胞を受容した細胞に機能分子を受け渡すことで、細胞間コミュニケーションツールとして様々な生命現象及び疾患に関与することが明らかになってきている。このような細胞外小胞(Extracellular vesicles : EVs)は、その産生経路もしくはサイズによって、エンドソームに由来する約100 nm のエクソソーム(Exosome)、細胞膜に由来するマイクロサイズのマイクロベシクル(Microvesicle)、死細胞の膜に由来するアポトーシス小体/小胞(Apoptotic body)の3種類に大きく分別できる。
細胞外小胞、特にエクソソームは、細胞、組織・臓器に対して様々な作用をもたらすと考えられており、その中でも抗炎症作用や免疫制御作用、組織修復作用などは特定の疾患に対する治療薬として活用できることが期待されている。特に、間葉系幹細胞(Mesenchymal stem cell : MSC)を用いた再生医療において、MSC が持つ治療効果は細胞自身が組織の細胞へ分化して修復する場合もあるが、MSC の分泌物(Secretome)による効果が高いとも考えられている1)。
このように、基礎研究が進みつつあるエクソソームに対して、製造工程において細胞加工物との類似性もあるため、再生医療等安全性確保法の対象に加えることが第69回厚生科学審議会再生医療等評価部会(令和3年12月1日)において議論された。結論は、細胞加工物というよりも細胞断片として扱い、最終的なヒトへの投与物としての明確な定義付けを行うことは、現時点では困難と判断された2)。今後の技術進展や諸外国における規制状況などの変化を踏まえて、再考される機会があると思われる。
研究開発の促進や、将来的に必要となる規制当局との対話の効率化など、エクソソームの産業化を実現させるための施策として、ガイドラインの整備は必須である。本稿では、主たる既存のガイドラインを概説したうえで、エクソソームへの適用可否、新たなガイドラインとして「標準」の必要性を説明する。
再生医療分野の標準開発状況
ICH(医薬品規制調和国際会議)では、医薬品の品質・有効性・安全性に関するガイドラインが整備されている(表1)。

しかしながら、これらは低分子医薬品、生物薬品を主な対象としたものであり、適用範囲に細胞は含まれていない。細胞は低分子化合物やタンパク質と比較して、化学式で構造を表現できない、サイズが桁違いで大きい、ヘテロ性が高いなど複雑であるため、これらのガイドラインを解釈して適用することは困難である。そこで、細胞を対象とした標準の整備が、ISO(国際標準化機構)を中心に進められている。標準は、開発プロセスに規制当局の承認が含まれない点を除き、ICHガイドラインと同義である。また、通常は3年と比較的短期間で開発できること、5年ごとに維持・改訂・廃止を判断する定期見直しを必須としていることから、先端分野のガイドラインとして適している。米国では、2016年12月に"21st Century Cures Act"が制定され、その3036項に、再生医療の開発、評価、及び審査を支援するために、アメリカ食品医薬品局(FDA)がアメリカ国立標準技術研究(NIST)などの利害関係者と標準開発の優先順位付けを行い、標準開発を推進することが規定されている4)。これを受けてFDAは、2019年3月に発出した産業界向けのガイダンスの中で、製品開発や承認申請に標準を活用するとそれらのプロセスが効率化されて成功確率が高まる、と説明して標準の活用を推奨している5)。
再生医療分野の国際標準は、国際標準化機構の専門委員会276(ISO/TC 276;バイオテクノロジー)において開発されている。本委員会は、2023年8月時点で4つの作業グループ(WG)から構成され、再生医療分野の標準は主として作業グループ3(WG 3;分析方法)及び作業グループ4(WG 4;バイオプロセッシング)において取り扱われている。また、36か国の積極参加メンバー(P-メンバー)と16か国のオブザーバーメンバー(O-メンバー)が参画(図1)、議長及び幹事をドイツ、WG 3のコンビーナを米国、WG 4のコンビーナを日本が担っている。
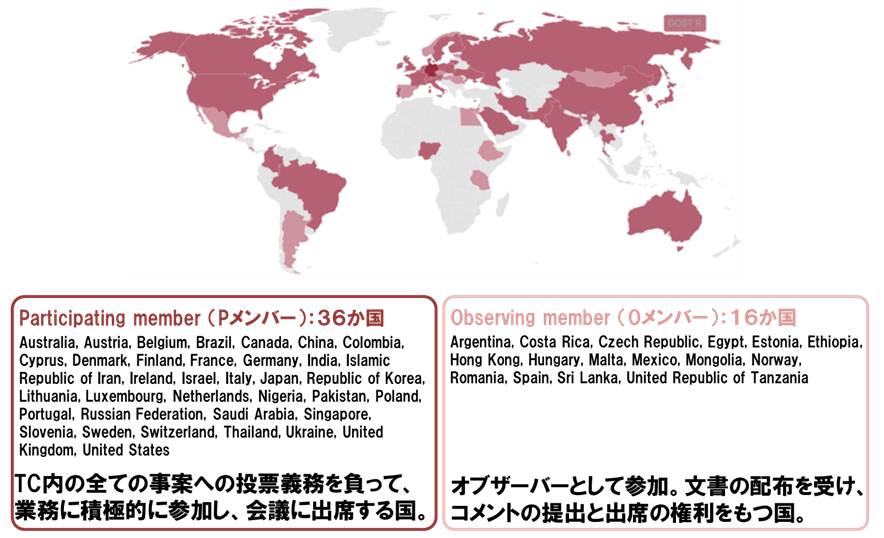
ISO/TC 276の国内審議団体は、日本産業標準調査会(JISC)から承認を受けた一般社団法人再生医療イノベーションフォーラム(FIRM)が担い、産官学より約85名が参画している。
ISO/TC 276において開発・発行されている再生医療に関わる標準を、「分析方法」、「周辺産業」、「製造」と分類して、以下で詳述する(図2、表2)。


「分析方法」に関する代表的な標準は、日米が共同開発したISO 23033(細胞治療製品の試験及び特性評価に関する一般要求事項及び考慮事項)6)であり、目的に適合する分析法のデザイン、分析法の妥当性確認、試験、及び報告に関する一般要求事項が規定されている。これは、ICH-Q6シリーズの細胞版と位置付けられ、従来は、各人がICH-Q6B(バイオ医薬品の規格及び試験方法)を参照して細胞を扱うための解釈を行い適用せざるを得ず、解釈の過程でバラツキが生じていた問題を解決した。
「周辺産業」に関する標準は、それぞれの対象とする製品・サービスの供給者と使用者(すなわち、細胞治療製品の開発・製造者)双方の役割を規定したものである。例えば、ISO 20399(細胞治療製品及び遺伝子治療製品の製造時に使用する補助材料)7)では、供給者が提供すべき情報及び使用者が採否を判断するために実施すべき事項を規定している。両者が標準に準拠することにより、使用者は目的に適合した補助材料の採用判断が可能となる。
「製造」に関する標準は、製品ライフサイクルを通して一貫性のある製造の実現を図るための「細胞製造マネジメントシステム」である。製造フローにおける不安定要因の抽出とリスクアセスメント、管理計画の策定、製造試作、レビューの実施、及び必要に応じこのサイクルを繰り返すPDCAを中心に規定したものである。日本が提案してアジア圏を中心に多くの国々の賛同を得つつあったが、英米の一方的な反対を受け、一旦提案を取り下げて、日本産業規格(JIS)としての開発を開始した。発行後に国内、次いでアジア圏に普及させ、再度ISOに提案する方針である。
エクソソームに対する標準活用・開発の提案
エクソソームは細胞を由来として製造されるため、生物薬品の一種と扱うことは可能である。しかし、タンパク質や核酸などを脂質二重膜が内包する多様な構成成分による複雑な構造物であるため、単純にタンパク質から成る抗体と同様に扱うことには難しさがある。エクソソームの製造プロセスに基づき、適用可能な標準、新規開発が必要な標準について考察する(図3)。

エクソソームの製造プロセスの概略は、細胞調製⇒細胞培養(エクソソーム産生)⇒濃縮・精製⇒製剤化である。 細胞調製及び培養工程においては、上記の再生医療に関わる標準群の適用が可能である。品質管理においては、ISO 23033をはじめとする「分析方法」に関わる標準群、製造関連では、ISO 20399、ISO/TS 23565などの「周辺産業」に関わる標準群、さらに「製造」に関わる細胞製造マネジメントシステムはそのまま適用可能である。ただし、細胞製造マネジメントシステムの目的であるQuality by Designの考え方の適用範囲を、エクソソームを含む培養液を最終製品と見做して培養工程までに留めるか、エクソソームの製剤化まで含む製造工程全体に広げるか、議論が必要である。
一方、工程モニタリングを含めた産生したエクソソームの分析には、ISO 23033にもICH-Q6Bにもエクソソームに特化した記述はなく、それらの適用は不十分である。エクソソームの特性解析と試験に関わる標準は、新規開発のニーズがある。
産生したエクソソームを含む細胞培養液を目的とする製品形態にするためには、濃縮やマイクロベシクル・アポトーシス小体・細胞や目的外のエクソソームを除去する精製が必要である。濃縮・精製工程に関しては、多くの方法が提案されているが、それらの中から方法を選択し、目的に適合する製品形態にすることは、エクソソームの産業化における鍵となる。論文などでは、超遠心法などによる濃縮を実施している例が多いが、多量処理ができないため、そのまま実用化に向けて適用することは難しい。また、精製方法によってはエクソソーム以外の不純物の混入が多く、精製度が最終製品の特性に影響するリスクがある。さらにエクソソームのサイズによる活性成分の違いや特定のサイズ以外の細胞外小胞が有効成分を含まない場合、それは不純物として捉えることもできるため、サイズの確認も重要になり得る。目的に適合した精製方法を選択することを支援するために、各精製方法の特徴、採用に当たって留意すべき事項などが記載されたガイドラインが必要となる。しかしながら、現状は適切なものが存在しておらず、新規標準開発のニーズとしてISO/TC 276/WG 4で議論が開始された。今後の進展に期待したい。
おわりに
基礎研究が着実に進展しているエクソソームの産業化に向けての課題の一つに、ガイドラインとして標準の整備が挙げられる。この目的に対し、再生医療用途として開発された標準群の活用の啓蒙、特性解析・試験、濃縮・精製を含むエクソソーム特有の課題に対処すべき標準開発の推進に取り組む。
参考文献
- エクソソームを含む細胞外小胞(EV)を利用した治療用製剤に関する報告書,独立行政法人医薬品医療機器総合機構
- 第69回厚生科学審議会再生医療等評価部会議事録
- ISO/IEC Guide 2 : 2004, Standardization and related activities -- General vocabulary(対応JIS;JIS Z 8002 : 2006, 標準化及び関連活動−一般的な用語)
- 21st Century Cures Act,
- Food and Drug Administration - Center for Biologics Evaluation and Research, Standards Development and the Use of Standards in Regulatory Submissions Reviewed in the Center for Biologics Evaluation and Research - Guidance for Industry, 2019
- ISO 23033 : 2021, Biotechnology -- Analytical methods -- General requirements and considerations for the testing and characterization of cellular therapeutic products
- ISO 20399 : 2022, Biotechnology -- Ancillary materials present during the production of cellular therapeutic products and gene therapy products
- ISO 20391-1 : 2018, Biotechnology -- Cell counting -- Part 1 : General guidance on cell counting methods
- ISO 20391-2 : 2019, Biotechnology -- Cell counting -- Part 2 : Experimental design and statistical analysis to quantify counting method performance
- ISO 24190 : 2023, Biotechnology -- Analytical methods -- Risk-based approach for method selection and validation for rapid microbial detection in bioprocesses
- ISO 20404 : 2023, Biotechnology -- Bioprocessing -- General requirements for the design of packaging to contain cells for therapeutic use
- ISO 21973 : 2020, Biotechnology -- General requirements for transportation of cells for therapeutic use
- ISO/TS 23565-1 : 2021, Biotechnology -- Bioprocessing -- General requirements and considerations for equipment systems used in the manufacturing of cells for therapeutic use
用語解説
ICHガイドライン
医薬品の品質・有効性・安全性の各分野のトピックごとに、各メンバーを代表する専門家が専門家作業部会で協議し、規制当局代表者の承認を経て作成される科学的・倫理的に適切と考えられる指針
[出典:PMDAホームページ(改変)]
標準
与えられた状況において最適な秩序を達成することを目的に、共通的に繰り返して使用するために、活動又はその結果に関する規則、指針又は特性を規定する文書であって、合意によって確立し、一般に認められている団体によって承認されているもの
[出典:JIS Z 8002,3.2]3)