【特別講座】ペプチド合成の最近の進歩
本記事は、OrganicSquare Vol.53 (2015年9月号)において、サイエンスライター 佐藤 健太郎 様に執筆いただいたものです。

プロテイン(protein)という言葉は、「第一の物質」を表すギリシャ語からきている。ペプチド及びタンパク質が、生命にとって文字通り「第一の物質」であることは、論をまたないところだろう。となれば、その合成法が化学における重要テーマであり続けていることも、不思議なことではない。
現在では、ほしい配列のタンパク質に対応する遺伝子を、大腸菌などの宿主に組み込んで発現させる遺伝子工学的な技法が普及し、十分な量のタンパク質を得ることが可能になっている。また近年では、大腸菌などを用いず酵素のみでタンパク質合成を行う、無細胞タンパク質合成系の研究も進んでいる。
とはいえこれらの方法では、基本的に 20 種の天然アミノ酸から成る単純なタンパク質しか合成できず、糖タンパク質やリン酸化タンパク質など重要な役割を担うタンパク質を得ることは難しい。また、非天然アミノ酸を含むタンパク質や、蛍光標識を組み込んだタンパク質なども、特殊な手法を用いない限り、今のところ合成困難だ。
一方、有機合成の技術でアミノ酸をつなぎ合わせる技法によれば、特殊アミノ酸も問題なくペプチド鎖に組み込める。また化学合成法では、洗練された技術がすでに蓄積されているため、大がかりな設備や熟達した技術者がなくとも、ペプチド合成が可能であることも魅力だ。
というわけで、フラスコ内でアミノ酸を連結させていく化学合成法も変わらず重要であり、一世紀以上の歴史を経た今も、なお研究の余地は多く残されている。その課題と最近の進歩について、簡単にまとめてみたい。
縮合
ペプチド合成の肝となるアミド結合を生成する手法については、多くの研究が積み重ねられてきた。この時問題となるのは、主にラセミ化(アミノ酸α炭素のエピ化)で、ペプチド合成において宿命としてつきまとう。特に、長いペプチド鎖同士を結合させる「フラグメント縮合」を行う場合、ラセミ化の問題は顕著となる。このためアミノ酸鎖伸長は、固相合成でも液相合成においても、N-保護アミノ酸を一つずつ N 端へ順次縮合させていくことが普通だ。
近年では、ラセミ化を抑制できる優秀な縮合剤が多数開発されている。たとえばよく用いられる縮合剤 O-(7-アザベンゾトリアゾール-1-イル)-N,N,N',N'-テトラメチルウロニウムヘキサフルオロホスファート(HATU)などは、ほとんどアミノ酸のラセミ化を伴わずカップリングが行える。最近登場した(1-シアノ-2-エトキシ-2-オキソエチリデンアミノオキシ)ジメチルアミノ-モルホリノ-カルベニウムヘキサフルオロリン酸塩(COMU)はさらに優秀で、フラグメント縮合においてもラセミ化を最小限に抑えられる 1)。
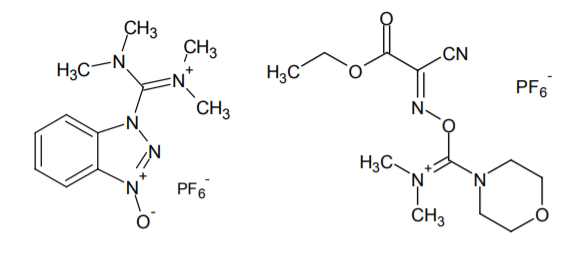
HATU(左)と COMU(右)
反応速度の面においても改善され、N-メチルアミノ酸やα,α-ジアルキルアミノ酸など、立体障害の大きなアミノ酸でも十分な速度でカップリングが行えるようになっている。また、水洗のみで副生成物を除去できるなど、使い勝手も優れている。これら縮合剤については、本誌 46 号(2013 年 12 月号)に詳述したので参考にされたい。
NCL 法の登場
通常行われる、1残基ずつアミノ酸鎖を伸ばしていく手法よりも、ある程度長いペプチド鎖同士の結合を繰り返す収束的な合成法のほうが、効率に勝ることはいうまでもない。ただし前述のように、この方法ではラセミ化という深刻な問題を伴う。
また、側鎖の保護されたペプチド鎖は溶解度に劣り、反応点同士が出会う確率も低くなるので、反応速度は一般に低下する。側鎖の保護されていないペプチド鎖同士を、選択的に N 端と C 端で結合させる方法があればよいが、長らくこれは夢のような話と考えられてきた。
1994 年、Kent らはこの壁を打ち破るネイティブ・ケミカル・ライゲーション法(NCL 法)を発表した 2)。突破口となったのは、システイン側鎖のメルカプト基の特殊な反応性であった。システインを N 末端に持ったペプチド鎖と、カルボン酸チオエステルとを反応させ、両者を結びつけるというものだ。
まずチオエステル交換反応によって、システインのメルカプト基がアシル化された中間体が生成し、次にアシル基が S→N 転位を起こして完全なペプチド鎖が得られる。この方法によれば、ペプチド鎖を傷める可能性のある酸・塩基・金属試薬などを用いることなく、全く無保護の状態のままで、ペプチド鎖同士を連結させることができる。つまり、遺伝子工学的手法で作ったペプチド及びタンパク質に対し、さらにアミノ酸鎖を延伸することも可能になる。
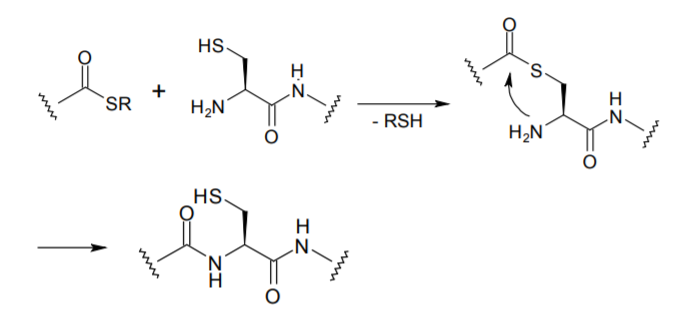
NCL 法によるアミド結合生成
この方法は、遺伝子工学的な手法では合成困難な糖タンパク質にも、適用が可能である。たとえば Bertozzi らは、この方法によって 82 残基から成るジプテリシンという抗菌性糖タンパクの改変体を合成した 3)。このペプチドは 10 番目と 54 番目のトレオニン残基に糖が結合しており、25 番目にシステイン残基を持つので、ここで NCL 法を適用することができる。
Bertozzi らはまず、1~24 番目と、25~82 番目のペプチドを別々に固相法で合成した。また 1~24 番目のペプチドは特殊なリンカーを介して樹脂と結合させてあり、ペプチド鎖を伸長させた後にベンジルチオールで処理すると、C 端がチオールエステルの形で得られる仕掛けがしてある。
こうして得られた両フラグメントを、pH7.5 のバッファ中室温で 18 時間反応させると、収率 55%で目的のペプチドが得られた。従来の液相法でも固相法でも、これだけのサイズとなると合成は難しいとされてきた。NCL 法の威力が、遺憾なく発揮されたといえる。
NCL 法の拡大
NCL 法の最も大きな弱点は、システイン残基のところでしか結合生成が行えない点だ。この制限があるため、システインが適切な位置にない、あるいはシステインを全く含まないペプチドやタンパク質に対しては、単純な NCL 法は適用できない。
そこで、いったんシステインの形でライゲーションを行った後、ラネーニッケルで処理することでシステインのメルカプト基を除去し、アラニンに変える方法が開発された 4)。また、N 端にペニシラミンを導入しておいてここでライゲーションを行い、同様に脱硫してバリンに変える手段も発表されている 5)。
アラニンやバリンはタンパク質中の含有比率が高いアミノ酸であるから、これによって NCL 法の適用範囲は大きく広がった。また Danishefsky らは、金属などを使わない脱硫方法を報告しており、多くの官能基を持つペプチドにも適用しやすい 6)。
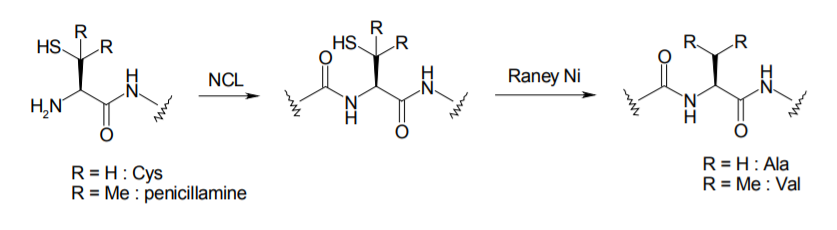
さらに長いペプチド鎖を合成したい場合には、1 ヶ所で NCL を行うだけでは不十分になる。そこで、N 端システインをチアゾリジンカルボン酸の形で「保護」しておくことにより、2 段階の NCL を行う手法も開発された。チアゾリジンカルボン酸はメトキシアミン塩酸塩などで処理すると、窒素とイオウを結ぶメチレンが脱離してシステインを遊離するので、ここでもう一段のライゲーションが可能になる。
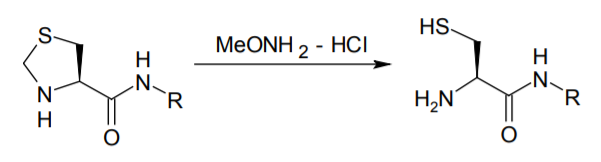
C. Unverzagt らはこの連続ライゲーションを用いて、124 残基のペプチド鎖に 9 糖から成る糖鎖が結合した、リボヌクレアーゼ C の合成に成功した 7)。40~124 番目に当たるペプチド鎖を遺伝子組み換え法によって作っておき、ここに固相法で合成した 26~39 番のペプチド(N 端はチアゾリジンカルボン酸、34 番のアスパラギンに糖鎖が結合している)を NCL で連結、メトキシアミン塩酸塩で処理した後 1~25 番のペプチドともう一度ライゲーションを行い、目的の糖タンパクを得ている。
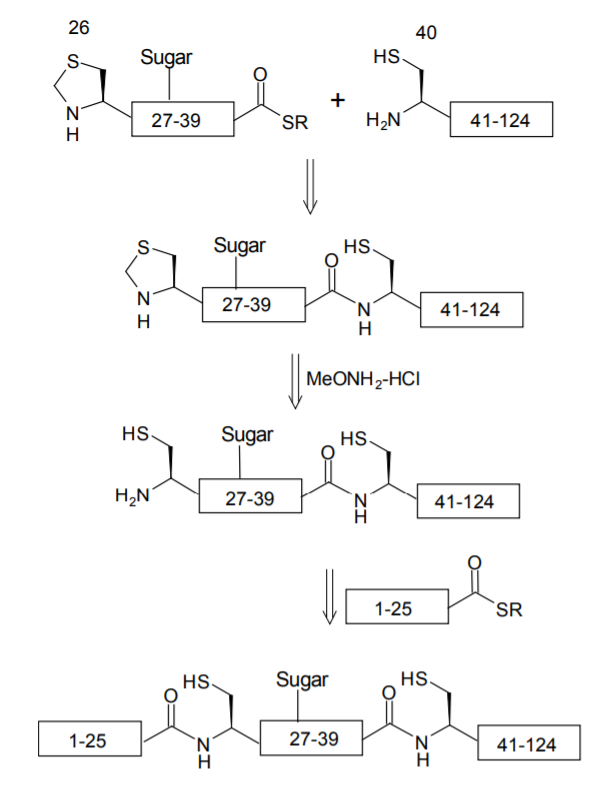
ほか、NCL 法にはさまざまなバリエーションが登場しており、適用範囲はますます広がっている。その他の方法については、総説を参照されたい 8)。
Bode 法
また Bode らは、これと異なるタイプのライゲーションを報告している。α-ケト酸と、N-ヒドロキシルアミンを単に混合するだけで、水と二酸化炭素の放出を伴いつつアミド結合が形成されるという方法である 9)。
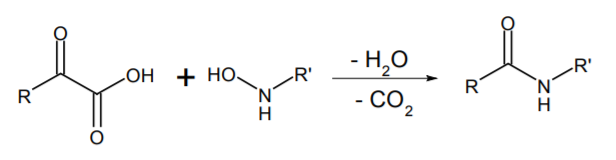
Bode 法は官能基許容性が高く、NCL 法と同様に側鎖は無保護のままでカップリング反応が行える。また、ペプチドの N 端アミノ基をヒドロキシルアミンへと酸化する反応 10)、C 端カルボキシ基からα-ケトカルボン酸へ変換する反応も報告 11)されており、多くの基質に適用できる条件が整えられている。
縮合剤などを全く用いない Bode 法は、ペプチド合成の歴史の中でも画期的といえる。副生成物は水と二酸化炭素だけなので、生きた細胞内などでの適用も可能だろう。さらなる展開が期待される。
マイクロフロー合成法
NMR や質量分析など、化学における分析手法はこの数十年の間に大きく進歩した。一方、反応を行う容器に関しては、フラスコと撹拌機の時代が長く続いており、100 年以上本質的に変わっていないともいえる。しかし近年になり、この分野にようやくマイクロフロー法と呼ばれる手法が、その一つだ。新しい波が訪れつつある。
マイクロフロー法は、細いチューブ状の空間に基質や試薬の溶液を送り込み、混合させて反応を行う仕組みであり、反応時間は混合液の流路の長さによって制御できる。反応容器として熱交換効率の高い素材を用いることで、精密な反応温度制御なども可能となっている。不安定な反応中間体を素早く次の反応に使用することで、通常低温条件を必要とする反応を、室温でも副反応なしに行なった例も報告されている。
このマイクロフロー法を、ペプチド合成に利用した例が登場してきた。東工大の布施らは、細いチューブ内で N-保護アミノ酸をトリホスゲンで活性化し、アミノ酸エステルと縮合させる反応を行った12)。この条件ではアミノ酸を酸塩化物として活性化するため、通常では反応性が高すぎ、かなりのラセミ化が起きてしまうのが常識であった。
布施らは反応条件を検討し、トリホスゲンによる活性化を 20℃、0.5 秒で、カップリングを 4.3 秒で行なった。この結果、ほとんどの基質でラセミ化は 1%以下に抑制され、90%以上の良好な収率でジペプチドが得られた。
温和な試薬で長時間かけて反応を行うのではなく、強い活性化法で瞬時に反応させるという発想の転換により、極めて高速に反応を行うことに成功した。トリホスゲンは他の縮合剤に比べて比較的安価であり、廃棄物もはるかに少なく済む。フロー時間を長くする、あるいは並行させて反応を行うことで、スケールアップも行いやすい。自動化にもつなげやすいから、将来応用範囲が広がることが期待される。
ペプチド合成は Fischer 以来の長い歴史を持つが、なお研究は積み重ねられ、革新的な技術も現れている。この結果、必ずしも有機合成に習熟していない他分野の研究者でも、十分利用可能な技術として洗練されつつある。また、この分野で得られた知見は、多くの有機合成反応に応用が可能だ。ペプチドを研究対象とする者のみならず、多くの科学者にとって目を向ける価値のある分野といえよう。
参考文献
- El-Faham, A., Albericio, F.: J. Org. Chem., 73, 2731 (2008). DOI: 10.1021/jo702622c
- Dawson, P. E. et al.: Science, 266, 776(1994). DOI: 10.1126/science.7973629
- Winans K. A. et al.: Biochemistry., 38, 11700 (1999). DOI: 10.1021/bi991247f
- Yan, L. Z. et al.: J. Am. Chem. Soc., 123, 526 (2001). DOI: 10.1021/ja003265m
- Haase, C. et al.: Angew. Chem. Int. Ed., 47, 6807 (2008). DOI: 10.1002/anie.200801590
- Wan, Q., Danishefsky, S.J.: Angew. Chem. Int. Ed., 46, 9248 (2007). DOI: 10.1002/anie.200704195
- Piontek, C. et al.: Angew. Chem. Int. Ed., 48, 1941 (2009). DOI: 10.1002/anie.200804735
- P. Thapa et al.: Molecules, 19, 14461(2014). DOI: 10.3390/molecules190914461
- Bode, J. W. et al.: Angew. Chem. Int. Ed., 45, 1248 (2006). DOI: 10.1002/anie.200503991
- Fukuzumi, T., Bode, J. W.: J. Am. Chem. Soc., 131, 3864 (2009). DOI: 10.1021/ja900601c
- Lei, J. et al.: J. Am. Chem. Soc., 130, 4253 (2008). DOI: 10.1021/ja800053t
- S. Fuse et al.: Angew. Chem. Int. Ed., 53, 851 (2014). DOI: 10.1002/anie.201307987
