Vol. 1. Overview of Alzheimer's Disease
This article was written by Dr. Ryoko Ihara, Department of Neuropathology, Graduate School of Medicine, The University of Tokyo, Unit for Early and Exploratory Clinical Development, The University of Tokyo Hospital.
Introduction
Alzheimer's disease (AD) is a neurodegenerative disease pathologically characterized by neuronal loss with senile plaques and neurofibrillary tangles predominantly in the hippocampus and the temporal lobe that clinically manifests as slowly progressive dementia primarily consisting of memory impairment in old age. There is not always a one-to-one correspondence between pathology and clinical presentation of AD, as evidenced by the fact that dementia, which primarily consists of memory impairment, is also caused by pathologies other than AD, these pathologies often coexist, and pure AD pathology is sometimes associated with atypical clinical symptoms. This article explains the molecular pathogenesis of AD as a neuropathological disease and then new clinical diagnostic criteria based on the molecular pathogenesis and the latest clinical studies.
Pathophysiology of AD
· Neuropathological findings of AD
In 1906, Alois Alzheimer reported that a microscopic examination of the brain from a patient with presenile dementia mainly characterized by memory impairment and delusion revealed senile plaques and neurofibrillary tangles, which were well demonstrated with the Bielschowsky silver stain. While senile plaques occur extracellularly, neurofibrillary tangles are intracellular aggregates (Fig. 1). A disease with these two pathological hallmarks was named AD separately from other dementias. Senile plaques and neurofibrillary tangles independently show relatively uniform pathological progression; more specifically, senile plaques progress from the cerebral neocortex to the mesial temporal lobe structures and basal ganglia, then to the brainstem and finally to the cerebellum,1) whereas neurofibrillary tangles progress from the entorhinal cortex to the limbic system and finally to the neocortex.2) In the National Institute on Aging-Alzheimer's Association (NIA-AA) guidelines for the neuropathologic assessment of Alzheimer's disease published in 2012, senile plaques and neurofibrillary tangles are graded according to the distribution and density to determine the plausibility of AD as the pathology responsible for dementia on a four-point scale.3)
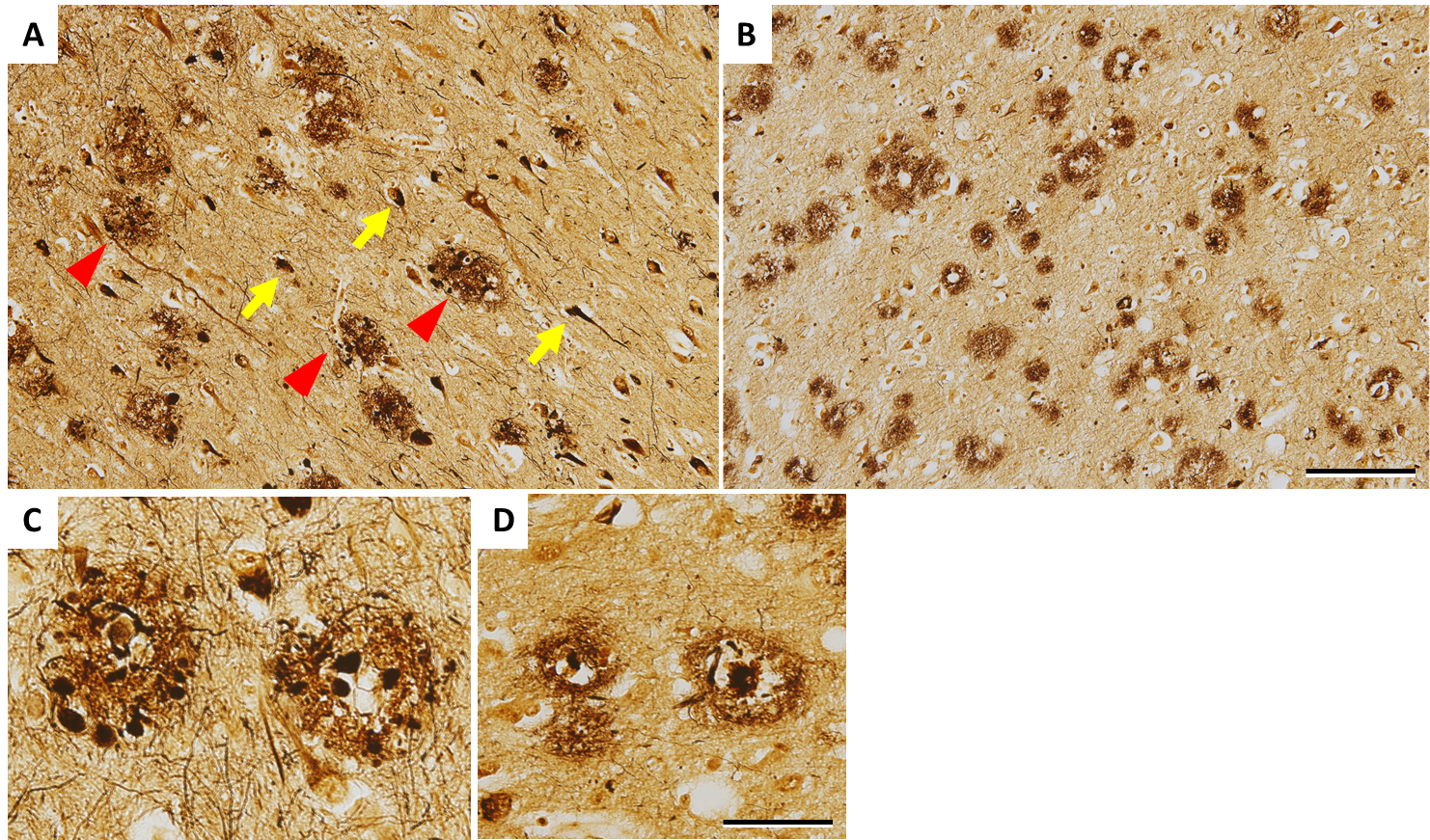
Figure 1. Histopathology of the brain from a patient with dominantly inherited AD (50s, PSEN1 V717I) A and C show the hippocampus, and B and D show the cortex of the temporal lobe, stained with the Bielschowsky silver stain. A: There are many senile plaques (arrowheads) and neurofibrillary tangles (arrows). B: There are senile plaques across the layer. C: There are senile plaques surrounded with dystrophic neurites ('neuritic plaques') in the hippocampus. D: A senile plaque with a core at the center as shown in this picture is called cored plaque. Scale bar: 100 μm in A and B, 50 μm in C and D
· Amyloid-β, tau, and amyloid cascade hypothesis
Due to the advances of biochemical techniques in the 1980s, it was successively reported that the main component of extracellular senile plaques was amyloid-β (Aβ), and the main component of intracellular neurofibrillary tangles was abnormally hyperphosphorylated tau protein, providing a breakthrough in research on the pathogenesis of AD. However, which of the two molecules characterizing AD plays a central role in the pathogenesis was debated for many years. In the 1990s, APP, PSEN1, and PSEN2 were successively identified as the genes responsible for autosomal-dominantly inherited AD, which accounts for approximately <1% of AD.4-6) Amyloid precursor protein, the product of APP, is a single-pass transmembrane protein, cleaved by the β-secretase beta-site APP cleaving enzyme (BACE1) at the luminal side and by γ-secretase within the plasma membrane into Aβ, which is then secreted extracellularly. Presenilin 1 and 2, the products of PSEN1 and PSEN2, respectively, constitute γ-secretase complex together with nicastrin, Aph-1, and Pen-2 to form the active center of the enzyme. All of the three causative genes are therefore related to Aβ production, whereas mutations in MAPT encoding tau protein result in not AD, but frontotemporal lobar degeneration; in addition, neuropathological analyses of brains from the cognitively unimpaired elderly exhibited that not a few individuals had many senile plaques, but no neurofibrillary tangles, leading to the belief that Aβ may be in the upstream of the pathogenesis of AD. The summarizing amyloid cascade hypothesis (also known as 'amyloid hypothesis' for short) assumes the pathological pathway as follows. First, mutations in APP, PSEN1, and PSEN2 result in increased Aβ production and secretion; second, Aβ oligomers are formed in the process of aggregation from Aβ monomers to senile plaques; third, the oligomers, which have synaptic toxicity, damage synapses and neurons; fourth, changes in kinase activity in neurons result in tau phosphorylation, which in turn results in neurofibrillary tangle formation; and finally, extensive neuronal dysfunction and neuronal death occur, leading to the clinical onset of cognitive dysfunction (Fig. 2).7) With multiple studies providing supportive evidence, for example, that tau knockout prevents cognitive dysfunction in APP transgenic mice, an AD model,8) and that APP mutations conferring resistance to β-secretase-mediated cleavage are associated with reduced risk for developing AD,9) the amyloid hypothesis, though hypothetical, is widely believed, while some findings require careful interpretation, including possible involvement of delayed degradation of extracellular Aβ, not overproduction of Aβ in pathogenesis of sporadic AD, and strong correlation of cognitive dysfunction with the progression of tau pathology rather than that of Aβ pathology. Disease-modifying drugs are currently being developed based on this hypothesis as the theoretical rationale, and the clinical diagnostic criteria using biomarkers described below are also determined on the pathological course assumed from the hypothesis.
The linkage between Aβ oligomer formation and tau-mediated abnormality, which is not thoroughly addressed in the hypothesis, remains the biggest black box, and the process in which APOE, the major risk gene for AD, is primarily involved still remains to be elucidated.
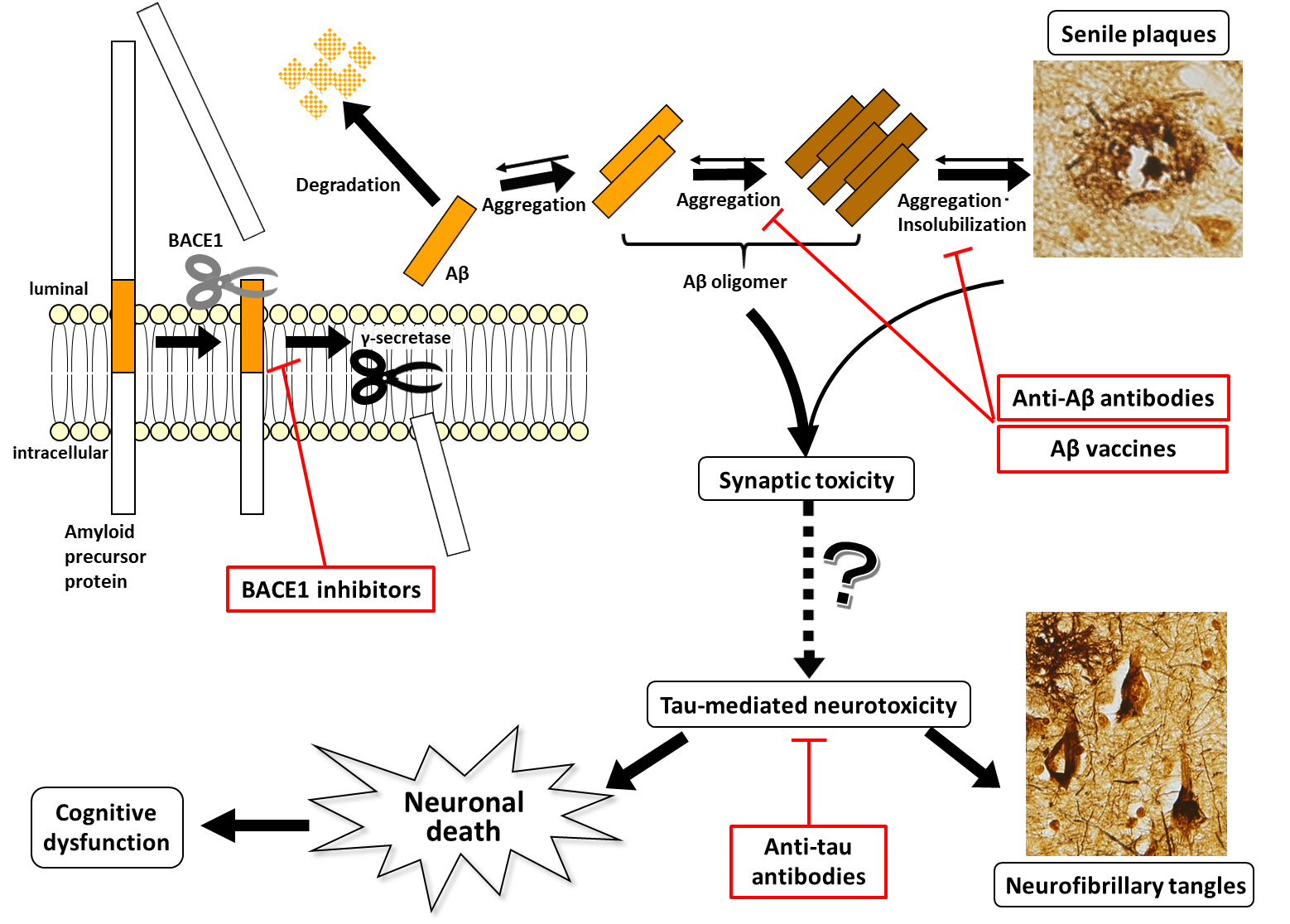
Figure 2. Amyloid hypothesis and disease-modifying drugs currently under development The amyloid hypothesis assumes that Aβ production results in the formation of oligomers, which have synaptic toxicity, resulting in tau-mediated neurotoxicity through an unknown mechanism and thereby leading to neuronal death. As shown in this figure, senile plaques and neurofibrillary tangles themselves may rather protect synapses and neurons from the toxicity of soluble forms of Aβ and tau by forming insoluble aggregates. Anti-Aβ antibodies act on various Aβ species that differ in the degree of aggregation.
· Biomarkers of AD
Biomarkers are quantitative measures reflecting the pathophysiology of a disease and used to diagnose the disease or assess its severity and progression. While scales for cognitive function and global function have been used as endpoints in clinical trials of drugs for AD, more quantitative biomarkers have been explored as alternatives to these scales which are substantially affected by internal and external factors. AD biomarkers which become detectable in the earliest stage are cerebrospinal fluid (CSF) Aβ (especially Aβ1-42) and amyloid PET, reflecting Aβ deposition in the brain, followed by FDG-PET and functional MRI, which reflect synaptic or neuronal dysfunction, CSF phosphorylated tau and tau and tau PET, which reflect tau-induced neurodegeneration, and structural MRI, which reflects neuronal death.10) Due to the recent remarkable progress, biomolecular imaging such as amyloid PET and tau PET have played an increasing role in clinical studies, including the use as eligibility criteria and secondary endpoints. Plasma or serum biomarkers have been desired as non-invasive and low-cost method for a decade, and they have been recently developed by using immunoprecipitated mass spectrometry or ultrasensitive immunoassay. Further validation for the new techniques is now ongoing.
Clinical diagnostic criteria of AD proposed by the NIA-AA
Dementia mainly characterized by memory impairment and suspected to be due to AD was called dementia of the Alzheimer type to distinguish it from pathological AD. However, more appropriate expression was desired due to the spread of the term "mild cognitive impairment (MCI)" as the prodromal stage of dementia since around 1995 as well as the biomarker-based evidence that clinical symptoms are notably preceded by pathological changes. In fact, the Dominantly Inherited Alzheimer Network (DIAN), a multicenter longitudinal observational study to follow up asymptomatic carriers of mutations causative for dominantly inherited AD, showed amyloid accumulation revealed by amyloid PET 20 years earlier than the onset of clinical symptoms.11) In these circumstances, the National Institute on Aging (NIA) and the Alzheimer's Association (AA) cooperatively announced new diagnostic criteria incorporating the latest biomarker findings in 2011.12-14) Twenty-five years had passed since the publication of the last version. According to the new diagnostic criteria, AD is classified into three clinical stages: AD dementia, MCI due to AD, and preclinical AD. Like other dementing illnesses, dementia is distinguished from MCI by impaired independent living, in addition to cognitive decline, and MCI is distinguished from cognitively unimpaired condition by consistent and often objective cognitive decline in daily life. On the other hand, biomarker information is added to define each diagnostic category more reliably. As concerns AD dementia, probable AD dementia can be diagnosed based only on clinical information that the disease is slowly progressive and characterized by memory impairment, impaired language function, impaired visuospatial abilities, and/or impaired executive function with objective evidence of deteriorating cognitive function, but can be diagnosed more reliably if positive for AD biomarkers. Similarly, MCI that is indicated not only by clinical information but also by biomarkers positive for both Aβ and neurodegeneration is distinguished as MCI due to AD - high likelihood. Preclinical AD is a clinically unimpaired condition or having subtle cognitive decline that is too slight to be diagnosed as MCI, but is diagnosed if positive for AD biomarkers. Assuming that AD biomarker which changes in the earliest stage is Aβ, followed by tau or neurodegeneration based on the aforementioned amyloid hypothesis, preclinical AD is classified into three stages according to the degree of progression: stage 1 if positive only for Aβ biomarkers; stage 2 if positive not only for Aβ biomarkers, but also for tau biomarkers or biomarkers indicative of neurodegeneration; and stage 3 if positive for both biomarkers with slightly reduced cognitive function. The NIA-AA diagnostic criteria, which are merely the consensus from multiple studies that biomarkers can be incorporated to make an antemortem pathological diagnosis precisely, should be helpful in selecting participants for observational studies or clinical trials, because definitive diagnostic categories are established for the purpose of research use. On the other hand, it is not recommended to measure biomarkers aggressively, but it is recommended to diagnose preclinical AD or measure biomarkers only for research purposes. In 2018, the NIA and AA further created a new research framework to define AD solely as a biological construct not as a clinical syndrome based on biomarkers.15)
Development of disease-modifying drugs for AD
Since Aricept was approved by the US Food and Drug Administration (FDA) for the treatment of AD dementia in 1996, only symptom-relieving drugs have been launched to the market so far. While these drugs are widely used in daily clinical practice, disease-modifying drugs that stop or delay the pathological process itself based on the amyloid hypothesis are awaited. Efforts have been made to develop disease-modifying drugs for nearly 20 years, but none have met primary endpoints in phase III studies.
The first candidate for clinical development was γ-secretase inhibitors that inhibit Aβ production, but the development of semagacestat, the first γ-secretase inhibitor, was discontinued due to an increased risk of skin cancer observed in its phase III study.16) Since this side effect is considered to be mediated by Notch, one of numerous substrates of γ-secretase, the development of Notch sparing γ-secretase inhibitors that do not inhibit Notch cleavage or γ-secretase modulators that inhibit the production of highly toxic Aβ42 alone without affecting γ-secretase activity was started, but discontinued or suspended due to failure to completely prevent the Notch-mediated side effects. Alternatively, BACE1 inhibitors that inhibit Aβ production at the first step have gained increasing interest. The clinical development of the first two BACE1 inhibitors was discontinued due to toxicity detected in non-clinical studies, but multiple BACE1 inhibitors are currently under clinical development. Given retinal lesions in BACE1 knockout mice,17) however, there is now a more cautious attitude towards the safety of inhibition of BACE1, which was previously considered a safer target than γ secretase, for which the existence of many substrates poses safety concerns. Furthermore, it was reported that multiple BACE1 inhibitors unexpectedly induced cognitive worsening in phase III studies in 2018-2019.18) Further laboratory experiments are needed to elucidate molecular mechanism of this critical side effect, and how to safely use BACE1 inhibitors and what stage of the disease BACE1 inhibitors will most effectively works are being discussed.
To promote Aβ clearance, anti-Aβ antibodies, which are passive immunization, and Aβ vaccines, which are active immunization, have been under clinical development. The first two anti-Aβ antibodies tested in clinical trials caused an adverse reaction of vasogenic edema known as amyloid-related imaging abnormalities (ARIAs), resulting in a temporal suspension of clinical development. Both antibodies underwent phase III trials through means such as dose reduction, but failed to prevent the progression of cognitive decline.19, 20) Solanezumab continued to be tested in clinical trials in MCI to early AD dementia, but was concluded to fail to achieve the endpoint in November 2016. Solanezumab is still being tested in clinical trials in preclinical AD described below. At present, multiple antibody pharmaceuticals targeting different Aβ species or different epitopes are under clinical development and appear to be promising; however, since ARIAs have to be prevented in dose selection, it is critical to appropriately select effective doses. The development of Aβ vaccines started earlier than that of antibodies, and AN1792, a full-length Aβ vaccine, was tested in a clinical trial in as early as 2001, but the development was discontinued due to meningoencephalitis reported as an adverse drug reaction.21) A less immunogenic vaccine against the N-terminal of Aβ is currently being tested in a phase II/III study.
Compounds targeting tau have also been under clinical development, but none have been demonstrated to be effective in phase III studies. Given the hypothesis that the pathological progression of tau may be triggered by prion-like propagation, in which extracellular pathological tau converts normal tau into pathological tau when taken up by neurons,22) anti-tau antibodies that inhibit the prion-like propagation are currently under clinical development, attracting interest.
New approach -Clinical trials in preclinical AD
Only a few researchers blame the falseness of the amyloid hypothesis for the consecutive failures of clinical trials of the disease-modifying drugs developed based on the amyloid hypothesis. While there is no doubt that drugs remain to be potentially improved, many researchers attribute the failures primarily to failure to administer the drugs to appropriate subjects in previous clinical trials. Firstly, it is questionable whether selected participants truly had a pathology to be treated; therefore, biomarkers are implemented to select participants for almost all the clinical trials in the early stage of the disease nowadays. Secondly, targeted stage of the disease may be too neuropathologically advanced to inhibit the progression of the pathological process. While clinical trials of disease-modifying drugs have gradually come to target earlier stage, that is, mild AD in place of moderate AD and then MCI in place of mild AD, Aβ accumulation almost reaches the maximum level and substantial brain atrophy, in other words, neuronal death already occurs in MCI in which initial clinical symptom is presented, indicating that even earlier intervention at the stage of milder pathological change may be desirable. Clinical trials in preclinical AD have been successively started since 2013, including the DIAN-trial unit (DIAN-TU) and the Alzheimer's Prevention Initiative (API) in asymptomatic carriers of mutations causative for dominantly inherited AD, and the Anti-Amyloid treatment in Asymptomatic Alzheimer's study (A4 study) in amyloid PET-positive sporadic preclinical AD. All these trials have a longer follow-up period (4 to 5 years) than traditional clinical trials due to the limited sample size for the DIAN-TU and the API as well as much smaller changes in cognitive function in the preclinical stage despite the acceptability of only cognitive measures as primary endpoints. These innovative clinical trials attract great interest from researchers and the general population, despite many challenges due to their being pioneer in the field, including huge costs, strategy required to reduce burden of participation on young participants, and cognitive measures to be refined to detect slight cognitive decline.
The latest keywords in clinical research in AD include public private partnership, a new framework to conduct these clinical trials, data sharing for efficient use of data from multicenter studies, and disease progression modeling, which analyzes the data and helps in designing efficient clinical trials.
Conclusion
This article explained the latest clinical studies while tracing the history of research on AD. In the past 40 years, many researchers have actively engaged in research on AD, successfully identifying the main components of pathological hallmarks and the genes causative for dominantly inherited AD, which led to elucidation of the pathogenesis and thus development of not only disease-modifying drugs but also clinically useful tools such as biomarkers and diagnostic criteria. Based on the foundation of longitudinal clinical data from cohorts including healthy individuals and autopsy findings accumulated over the same period of time, on the other hand, a new form of clinical studies combining basic research with clinical research are being accumulated. There is no doubt that the history of research on AD will serve as a model for the development of treatments for other neurodegenerative diseases.
References
- Thal, D. R. et al . : Neurology , 58, 1791-800(2002)
- Braak, H. et al . : Acta Neuropathol ., 112, 389-404(2006)
- Montine, T. J. et al . ; National Institute on Aging ; Alzheimer's Association : Acta Neuropathol ., 123, 1-11(2002)
- Goate, A. et al . : Nature , 349, 704-6(1991)
- Sherrington, R. et al . : Nature , 375, 754-60(1995)
- Levy-Lahad, E. et al . : Science , 269, 970-3(1995)
- Hardy, J. et al . : Science , 297, 353-6(2002)
- Roberson, E. D. et al . : Science , 316, 750-4(2007)
- Jonsson, T. et al . : Nature , 488, 96-9(2012)
- Jack, C. R. Jr. et al . : Lancet Neurol ., 9, 119-28(2010)
- Bateman, R. J. et al . : Dominantly Inherited Alzheimer Network : N. Engl. J. Med ., 367, 795-804(2012)
- McKhann, G. M. et al . : Alzheimers Dement .,7, 263-9(2011)
- Albert, M. S. et al . : Alzheimers Dement ., 7,270-9(2011)
- Sperling, R. A. et al . : Alzheimers Dement .,7, 280-92(2011)
- Jack, C. R. et al . : Alzheimers Dement .,14, 535-62(2018)
- Doody, R. S. et al . ; Alzheimer's Disease Cooperative Study Steering Committee; Semagacestat Study Group : N. Engl. J. Med ., 369, 341-50(2013)
- Cai, J. et al . : EMBO Mol. Med ., 4, 980-91(2012)
- Egan, M. F. et al . : N. Engl. J. Med ., 380, 1408-20(2019)
- Salloway, S. et al . ; Bapineuzumab 301 and 302 Clinical Trial Investigators : N. Engl. J. Med ., 370, 322-33(2014)
- Doody, R. S. et al . ; Alzheimer's Disease Cooperative Study Steering Committee; Solanezumab Study Group : N. Engl. J. Med ., 370, 311-21(2014)
- Orgogozo, J. M. et al . : Neurology , 61, 46-54(2003)
- Holmes, B. B. et al . : J. Biol. Chem ., 289, 19855-61(2014)