Risk Assessment and Control in the Guideline for Elemental Impurities in Drugs
The original article by Rina Nishioka, Soichiro Hanakawa of the Sumika Chemical Analysis Service, Ltd., appeared in an e-newsletter of FUJIFILM Wako Pure Chemical Corporation and was translated by the company.
1. Introduction
A regulatory guideline for elemental impurities in drug products was formulated by the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). In Japan, it was published as "Note on the Guideline for Elemental Impurities in Pharmaceuticals" (Yakushokushinsahatsu Notification No. 0930-4 from the Evaluation and Licensing Division, Pharmaceutical and Food Safety Bureau, Ministry of Health, Labour and Welfare [MHLW], dated September 30, 2015). A Second Supplement to the Japanese Pharmacopoeia (JP) XVII (MHLW Notification No. 49, dated June 28, 2019), published a few years ago, includes the General Information "Control of elemental impurities in drug products" in line with the Guideline. Further, the JP XVIII, announced in June 2021, includes an additional provision to the GENERAL NOTICES for control of elemental impurities, and an amendment to the General Tests, Processes and Apparatus. The updates are summarized below.
Product risk assessment, an important task in the control of elemental impurities, and its procedures are then described briefly. An example approach to risk assessment is described below.
2. Updates in the JP XVIII
The JP XVIII newly incorporated the control of elemental purities under General Notice 34, requiring any JP-listed drug product to be subject to proper impurity control in accordance with the provisions concerning elemental impurities in the JP General Tests, Processes and Apparatus. Drug products, drug substances, and additives, and other items are required to meet the requirements on impurity control based on the Guideline within 36 months following the enforcement of the Notification (by June 2024).
Provisions for tests were developed by merging Section 2.66, "Elemental impurities" in the General Tests, Processes and Apparatus and "Control of elemental impurities in drug products" in the General Information. The content reflects the modifications of allowable daily exposure to chromium which appeared in the ICH Q3D "Note on the Amendment to the Guideline for Elemental Impurities in Pharmaceuticals (Yakuseiyakushinhatsu Notification No. 0626-1 from the Director of the Drugs Evaluation and Licensing Division, Pharmaceutical Safety and Environmental Health Bureau, MHLW, dated June 26, 2020).
Considering that 36 months have passed after the announcement of the Guideline, it has been decided that existing drug products are included in the scope of the Guideline. Evaluation and control of elemental impurities are expected to be increasingly emphasized.
The ICH elemental impurities Guideline is applicable to new drug products when new drug manufacture and marketing is applied, whereas drug products for prescription other than those listed in the JP are subject to similar control under the expanded scope of application of the present notification of the Guideline according to the "Note on the Handling of Elemental Impurities in Ethical Drugs" (Yakuseiyakushinhatsu Notification No. 1228-7, dated December 28, 2020).
3. Risk assessment
The elemental impurities risk assessment process is performed in the following steps:
Identification
Identify potential sources of elemental impurities and their way into drug products
Evaluation
Evaluate elemental impurities in the drug product through comparison of their observed or predicted values and permitted daily exposure (PDE) values
Summarization
The results of the risk assessment are summarized and documented. It is then considered whether control procedures are appropriate or whether any further control procedures are necessary.
These steps are iterated to establish the appropriate control procedures.
3.1. Identification of sources of elemental impurities
The followings are potential sources of contamination with elemental impurities to be considered in the manufacturing process for the drug product:
- Residual impurities from elements that have been intentionally added during the manufacture of drug product ingredients such as drug substances and excipients (e.g. catalysts)
- Elemental impurities that are not intentionally added to the drug substance, water, or excipients, and are potentially therein
- Elemental impurities that are potentially introduced into the drug substance and/or drug product from manufacturing equipment.
- Elemental impurities that have the potential to be leached into the drug substance and drug product from container closure systems.
Figure 1 shows the components of the manufacturing process for the drug product. In risk assessment, it must be taken into account that the amounts of elemental impurities from potential sources in each component can influence the total amount of elemental impurities in the drug product.
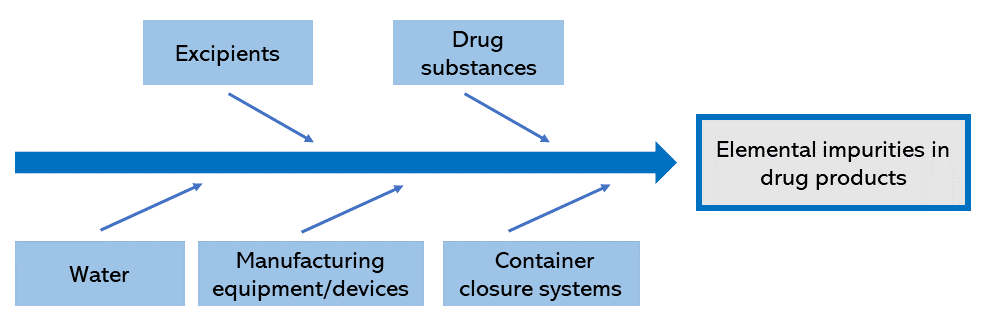 Figure 1. Potential sources of contamination with elemental impurities
Figure 1. Potential sources of contamination with elemental impurities
There are two common approaches to a risk assessment: the "component approach," which focuses on elemental impurities in various components, and the "drug product approach," which focuses on elemental impurities in the finished product. Sources of contamination and other factors to be considered in the approaches are shown in Figure 2. With regard to these sources of contamination, evidence data, such as published documents and information and test results from the respective suppliers, are obtained to identify impurities with high impact.
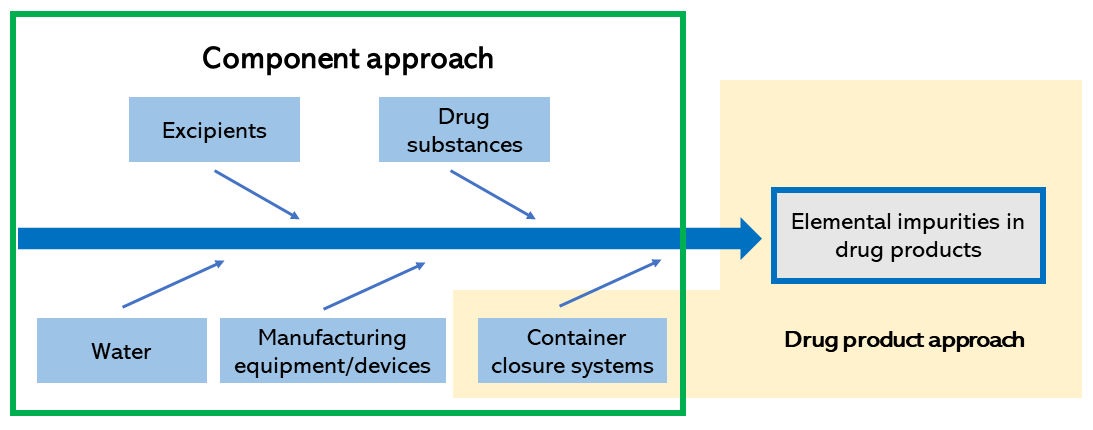 Figure 2. Two approaches to risk assessment
Figure 2. Two approaches to risk assessment
Note that the elemental impurities Guideline includes a training method consisting of 10 modules (0 to 9) to ensure further understanding. For the approach procedures, refer to the detailed information in Training Methods, Module 5.
3.2. Evaluation of elemental impurities (conversion between PDE values and maximum allowable concentration)
After identifying the source of contamination and target elements, individual elemental impurities in the drug product are evaluated by comparing observed or predicted values and preset PDE values. The Guideline describes four methods for converting the PDE values to the maximum allowable concentrations before comparing them with the observed or predicted values: Options 1, 2a, 2b, and 3. In the component approach, the appropriate method is selected from among Options 1, 2a, and 2b. In the drug product approach, Option 3 is used for the conversion to evaluate the finished product. (For details of the evaluation, refer to Section 7 of the Guideline.)
3.3. Control of elemental impurities
The Guideline defines the control threshold as 30% of the PDE value. If the level of any elemental impurity is lower than the control threshold, no additional control is required. If the level is likely to exceed the control threshold but is lower than the PDE value, it is necessary to take action to lower the level. Example countermeasures include upstream control, reselection of components and the container and closure system, and establishment of specifications.
If the level of impurity exceeds the PDE value, further countermeasures are required. However, if no technically feasible countermeasures are available, the level may be accepted under specific conditions such as shown in Training Methods, Module 6 by explaining rationale.
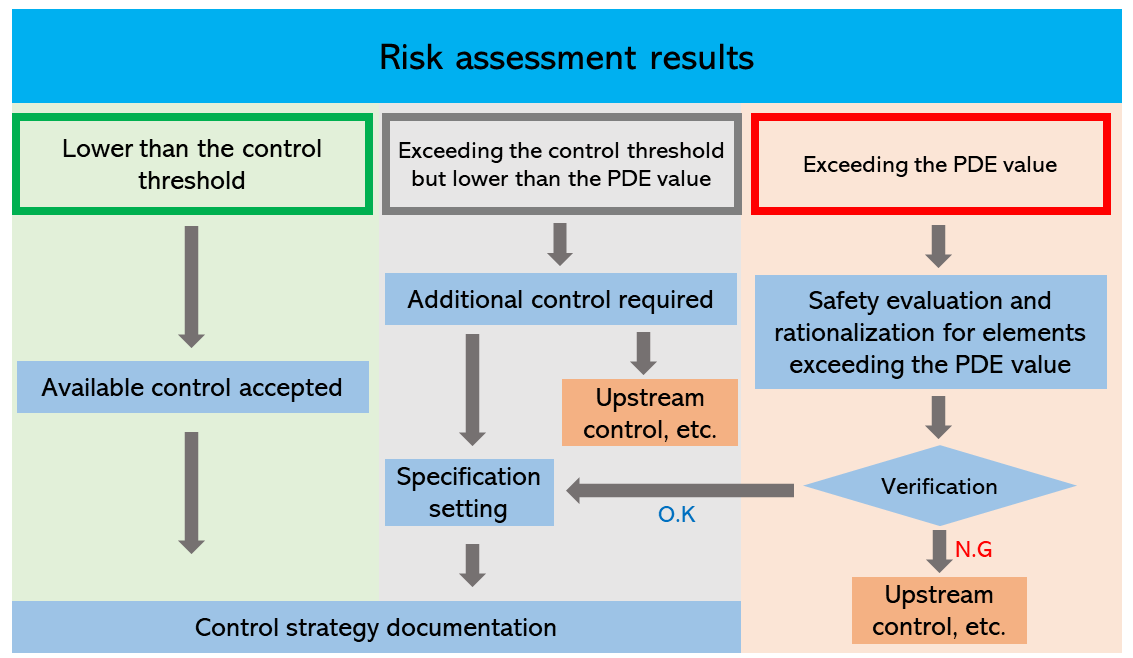 Figure 3. Diagram for control of elemental impurities
Figure 3. Diagram for control of elemental impurities
4. Primary evaluation by screening
Many methods of risk assessment are available. When using the drug product approach to perform the assessment, the first step is collecting quantitative data on elemental impurities in the drug product, and this is followed by identification of target elements to be assessed. If the available quantitative data on elemental impurities are insufficient, it is necessary to actually quantify them in the drug product. An effective method of narrowing target elements is to employ screening analysis to compare the maximum allowable concentration control thresholds for the 24 target elements listed in ICH Q3D.
Here, an imaginary approach to screening analysis for parenteral product is described below.
| Route of administration | Injection |
|---|---|
| Maximum daily intake | 2 mL |
| Target elements | Class 1, 2A, 2B, and 3 (24 elements) |
| Option | 3 |
| Limit of Quantitation | ≤10% of permitted daily exposure (PDE) |
| Instrument | ICP-MS |
Table 1 shows the permitted concentrations, control thresholds, limit of quantitation, and results for some of the target elements examined in the screening analysis.
Table 1. Permitted concentrations, control thresholds,
limit of quantitation, and results in the screening analysis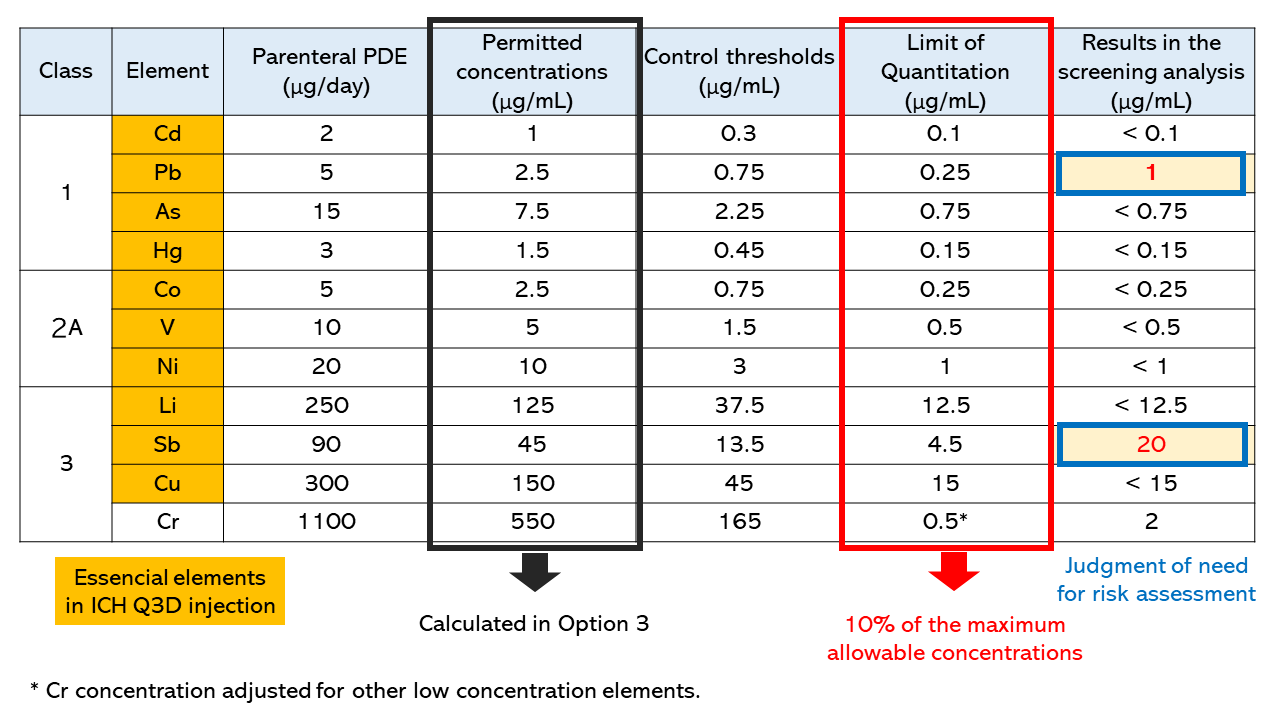
The permitted concentrations were calculated by dividing the parenteral PDE values by the maximum daily intake (2 mL) in Option 3. In this case, the limit of quantitation was set at 10% of the permitted concentrations or lower. For elements for which the permitted concentration is particularly higher than those of other elements, such as chromium, contamination with the elemental impurities can be checked by evaluating its limit of quantitation in the measurement at lower concentrations, as with other elements.
In addition, the permitted concentration varies among different target elements, as indicated in the example; therefore, it will become easier to prepare standard solutions by using commercially available mixed standard solutions whose concentrations are pre-adjusted to the ICH Q3D requirements to skip the dilution work and ensure high accuracy.
In this case, Pb and Sb were detected at levels higher than their respective control thresholds; therefore, additional control appears to be necessary, including the identification of sources of the contamination, upstream control, and, if necessary, the establishment of specifications for this product.
In the case of parenteral products, there are concerns not only on elemental impurities in the drug product, but also on the risk of leaching package material components during the shelf life of the drug product. The risk of contamination from the container and closure system must be separately evaluated on the basis of test data from stability testing and tests under severe conditions, or information collected from component manufacturers.
5. Conclusion
A screening evaluation to explain the content of risk assessment was described with parenteral product as an example. There are various cased for risk assessment of elemental impurities depending on the route of administration, components, and other factors, necessitating actions in line with different situations. It is recommended that the manufacturer refer not only to the Guideline, but also to other various sources of information, such as the Training Modules, that show details and literature data on excipients used outside Japan. It should be noted that, as described in Section 2, JP drug products and non-JP drug products are required to meet the requirements of control of elemental impurities based on the Guideline Notification and the provisions in the new pharmacopoeia, within 36 months after the enforcement of the official announcement of the JP XVIII.