Vol. 2. Tau protein in Alzheimer's disease
This article was written by Dr. Masato Hasegawa, Department of Dementia and Higher Brain Function, Tokyo Metropolitan Institute of Medical Science.
Introduction
Alzheimer's disease (AD) is neuropathologically characterized by two abnormal structures: senile plaques and neurofibrillary tangles. Senile plaques are extracellular structures composed of amyloid β (Αβ) peptides of ~42 amino acids, and neurofibrillary tangles are intracellular structures composed of microtubule-associated tau protein (tau). It has long been debated which of them is key to the onset and pathogenesis of Alzheimer's disease, and what is the relationship between the two pathologies. However, these questions remain open. The "amyloid hypothesis" has been widely accepted for the pathogenesis of AD since several mutations in the Αβ precursor protein (APP) gene were discovered in familial forms of AD (FAD) in 1990. Thus, many AD researchers and pharmaceutical companies have concentrated on studying the molecular mechanisms of Aβ production and aggregation, and have focused on developing drugs to inhibit APP cleavage and Aβ aggregation. However, all trials of AD therapies targeting Αβ have failed so far. On the other hand, tau pathologies are also one of the defining features of AD, as well as various other neurological dementing disorders, and therefore tau has recently attracted increasing attention as a therapeutic target of AD. Discoveries of mutations in the tau gene in frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17) have provided direct evidence that tau abnormalities cause tau accumulation and neurodegeneration rather than representing a final common pathway. Here, I will review tau pathologies in AD and other dementing disorders, and discuss the molecular mechanisms of pathogenesis and progression of tau-related diseases, focusing especially the prion-like propagation of pathological tau.
Structure, function and localization of tau protein
Tau is a microtubule-associated protein that is expressed in neuronal and glial cells. It is mainly localized in axons and functions to promote microtubule polymerization and stabilization. In adult human brain, six tau isoforms (352 to 441 amino acids) are produced by alternative splicing of mRNA transcribed from one tau gene (MAPT) in chromosome 17 (Fig. 1, A). In the central nervous system, the shortest 3-repeat tau isoform is expressed in the fetal to juvenile stages, and longer isoforms become expressed during development by insertion of exons 2, 3 and 10. Finally, all six tau isoforms are expressed in adult human brain, with apparent molecular weights of 48 ~ 67 kDa (Fig. 1, B) 1). The 3R tau and 4R tau proteins are produced in the absence or presence of exon 10 insertion, respectively. Since the repeat sequences function as microtubule binding domains, 4R tau isoforms have a higher microtubule polymerization ability than 3R tau isoforms. Although the expression ratio of 3R tau to 4R tau is approximately 1: 1 in the adult human brain, the ratio is different in animals. Further, if the ratio is affected by MAPT mutations, carriers will develop tauopathy as adults. In adult brains of rodents only 4-repeat tau isoforms are expressed, in contrast to human brain. Due to this difference in the tau isoforms, Alzheimer-type tau pathologies may not be reproduced in wild-type mice.
Tau was originally identified as a heat-stable microtubule polymerization-promoting factor, and is not denatured or inactivated even if treated at 100°C for 5 minutes 2). Its stability may be due to its natively unfolded state with little tertiary structure, which is rare in proteins with relatively high molecular weight. But, this property makes it possible to purify the protein relatively easily even from brain samples. Tau is a phospho protein and phosphorylation reduces its microtubule polymerization ability. Thus, phosphorylation may regulate tau binding to tubulin and microtubule polymerization. Tau is highly phosphorylated in brains at the juvenile stage (20 to 80% in the fetal period, and about 0 to 50% in adult stages 3)), when neurites and axons are extending and/or retracting repeatedly, although the proportions of phosphorylated form differ among sites.
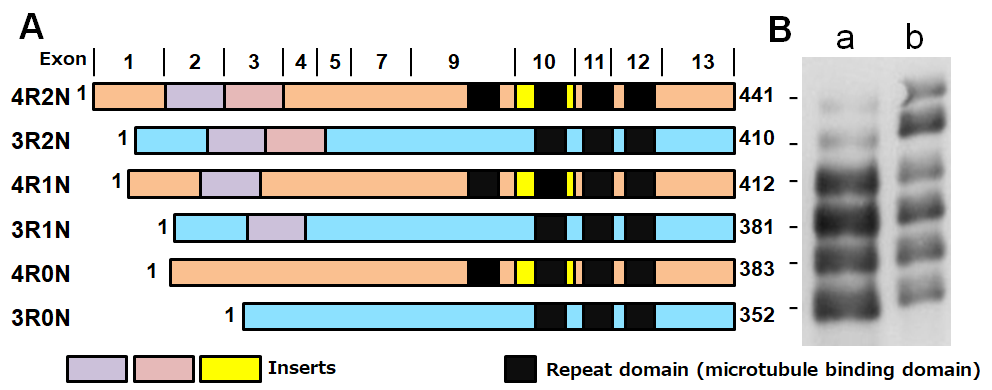
Figure1. Schematic diagrams of six tau isoforms in human brain
A. Schematic diagrams of six brain tau isoforms,
B. Gel electrophoresis of tau: a, soluble tau (after dephosphorylation), b. purified recombinant human tau proteins (immunoblotted with anti-tau antibody T46).
Pathological tau in AD
So, what kind of tau protein is deposited and how is it accumulated in brains of AD patients? As mentioned above, tau is accumulated as neuropathological inclusions called neurofibrillary tangles and neuropil threads (Fig 2A), which are characteristic lesions of AD. Electron microscopic observation shows that they have unique filamentous structures, so-called paired helical filaments (PHFs) and straight filaments (SFs)(Fig. 2B). Since 1990, biochemical and morphological analyses of tau in AD brains by various methods, including electron microscopy, immunochemical analyses with phosphorylation-specific antibodies, and mass spectrometric analysis, have clarified the structural and molecular features, as summarized in Table 1 4).
1) Normal tau is a highly soluble protein with little tertiary structure, whereas pathological tau has an amyloid-like fibril or filamentous form with cross-β structure, which is highly insoluble in detergents such as Triton-X and Sarkosyl. It is highly resistant to proteases, phosphatases, etc.
2) All six tau isoforms expressed in adult human brains are accumulated in AD brains, but they are much more highly phosphorylated than normal tau, both qualitatively and quantitatively. The hyperphosphorylated full-length tau isoforms are detected with phosphorylation-dependent and -independent anti-tau antibodies as triplet bands having apparent molecular weights of 60, 64, and 68 kDa (Fig. 2C).
3) Pathological tau in AD is partially fragmented, ubiquitinated, deamidated, and shows smearing in SDS-PAGE.
The next questions to arise are, what is the timing of the abnormal post-translational modifications in tau, and what is their relationship to tau accumulation? In the 1990's, attention was mainly focused on the abnormal phosphorylation of tau, and many reports on tau kinases and phosphatases appeared. It was probably easy to accept the idea that tau dissociates from microtubules upon phosphorylation and then self-aggregates, since signal transduction by phosphorylation was a common theme in cell biology at that time. Even now, many researchers support this idea. However, it is difficult to reproduce the abnormal phosphorylation state of AD-tau simply by phosphorylating tau using various kinases in the presence or absence of various phosphatase inhibitors. Based on our analyses of tau in AD brains and experimental models, it is reasonable to conclude that phosphorylation is not a cause, but rather may be a cellular response that occurs after aggregation or fibril formation of tau 4). Phosphorylation, ubiquitination, and fragmentation of tau might occur after aggregation as a kind of defensive reaction to degrade and eliminate abnormal protein aggregates in cells. Notably, filamentous forms of tau in AD cannot be dephosphorylated by protein phosphatases without denaturation: that is presumably why pathological tau proteins are resistant to dephosphorylation postmortem.

Table1. Comparison of normal tau and abnormal tau in AD
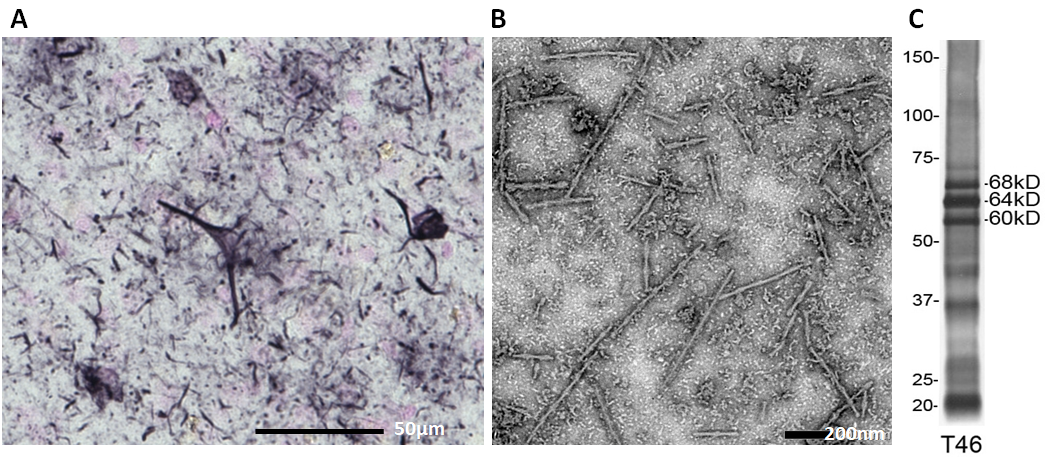
Figure 2.
A. Immunostaining of AD brain section with AT8. Neurofibrillary tangles in soma and neuropil threads in processes are stained.
B. EM picture of tau filaments in sarkosyl-insoluble fraction prepared from an AD brain. (negative staining).
C. Immunoblot analysis (with anti-tau T46) of sarkosyl-insoluble abnormal tau prepared from an AD brain. Triplet bands of 60, 64 and 68 kDa represent hyperphosphorylated full-length tau isoforms. C-Terminal fragments and smears are also detected.
Relationship between tau and Aβ in AD
As described above, tau proteins are deposited in AD brains as amyloid-like fibrils, which are positively stained by β-sheet ligands such as Congo red and thioflavin. The idea of seed-dependent aggregation is now generally accepted to explain the formation of amyloids or amyloid-like fibrils. First, aggregation nuclei are formed, then normal proteins binds to these seeds, and the fibrils are elongated. The initial seed may be generated as a result of aging, dysfunction, failures in protein degradation and quality control systems, and so on. It is important to elucidate the causes, but from the therapeutic point of view, it is more important to control the elongation process of abnormal tau, since considerable amounts of abnormal tau have already accumulated by the time symptoms appear. Based on their neuropathological analyses of many control and AD cases, Braak et al reported that tau lesions first appear in patients in their early 30's, prior to Aβ pathology. These lesions appear initially in the pre-hippocampal olfactory area (possibly originating from the locus coeruleus) and gradually spread to the limbic system, such as the hippocampus, the Meynert basal ganglia, and the amygdala, and then further to the neocortex5). Importantly, the distribution and spread of tau pathologies are well correlated with disease symptoms, and are used as a part of the Braak staging of AD progression. Recently, PET probes for Aβ and tau imaging have been developed, and so it has become possible to see where and how these pathologies are distributed in living patients. PET imaging should firmly underpin Braak staging in the near future. The stereotypic tau spreading in AD is well-known, but little attention was given to why and how the tau lesions spread during the course of the disease progression, although a viral infection was often assumed. More recently, it has been proposed that intracellular abnormal proteins, including tau, have prion-like properties, i.e., the abnormal protein can convert normal protein into abnormal form, which spreads from cell to cell in the same way as protein infectious particles known as prions 6).
Tau aggregation in other neurodegenerative diseases
Abnormal tau pathologies are formed not only in AD but also in other neurodegenerative diseases, such as Pick's disease (PiD), progressive supranuclear palsy (PSP) and cortical basal ganglia degeneration (CBD). Therefore, some researchers thought that tau aggregation might be a final common pathway of neurodegeneration, rather than the cause. However, in 1998, MAPT mutations were found in a familial form of dementia with tau pathologies (FTDP-17). The discovery of MAPT mutations in FTDP-17 indicated that genetic abnormalities induce tau aggregation and neurodegeneration, and abnormal tau is the cause of degeneration in those diseases. FTDP-17 is extremely diverse both clinically and neuropathologically, but can be classified according to the positions of tau mutations and the effects. For example, cases with R406W tau mutation develop neurofibrillary tangles like those in AD, while cases with N279K tau mutation show abundant glial tau pathologies in white matter. In vitro experiments indicate that many missense mutations affect the structure and function of tau, which may induce tau aggregation, while some mutations in exon 10 and introns affect the splicing of exon 10 and change the expression ratio of 3R tau to 4R tau. A common feature of FTDP-17 cases is tau accumulation, suggesting that neurodegeneration occurs via tau accumulation. Consequently, neurodegenerative diseases with tau pathologies are referred to as tauopathies. The tau pathologies, including accumulated tau isoforms, affected cell types and anatomical distributions, distinctly differentiate these diseases. In PiD, 3R tau isoforms accumulate in small neurons in the second layer of cortices, while in PSP and CBD, 4R tau isoforms accumulate in both neuronal and glial cells. On the other hand, in AD, both 3R and 4R tau isoforms accumulate in relatively large nerve (pyramidal) neurons with a ratio of 3R tau and 4R tau of approximately 1: 1. The ultrastructures of tau aggregates also differ among the diseases. Tau accumulates as straight filaments in PiD, but as twisted narrow and wide ribbon-like structures in PSP and CBD, respectively. In AD, both 3R and 4R tau accumulate with paired helical filament morphology. The morphological differences are reflected by biochemical analyses: the diseases can be classified according to the band pattern of the C-terminal fragments of tau and differences in protease-resistant bands7). Thus, the pathological tau proteins are characteristic defining features of these diseases. The data strongly suggest that they are the causes of neurodegeneration, and their accumulation is closely correlated with disease progression.
Experimental models of tau aggregation and propagation
How are such diverse tau pathologies formed? Many attempts have been made to reproduce the abnormal tau accumulation in cultured cells and mice, using overexpression models or mutant tau proteins. However, despite the observation of elevated levels of phosphorylated tau and mislocalation of tau in cell bodies and dendrites, accumulation of filamentous tau has hardly ever been detected, except in transgenic mice overexpressing P301L and P301S mutant tau. The major reason for this may be the time factor: abnormal tau pathology cannot be recapitulated within the short lifespan of mice. However, we considered that this could be overcome by introducing seeds into cells or by injecting seeds into the brain, as this by-passes the rate-limiting step of aggregation, going directly to the elongation process, so that aggregated protein pathologies can form very rapidly.
When we investigated the toxicity of tau aggregates, we fortunately found that tau fibrils could be efficiently introduced into cells by lipofection, and since then we have been working on seed-dependent aggregation of intracellular proteins. Tau fibrils served as seeds, and converted normal tau in cells to an abnormal form, resulting in the accumulation of phosphorylated tau. Interestingly, 3R tau was accumulated only when 3R tau fibrils were introduced, and 4R tau was accumulated only when 4R tau fibrils were introduced. Thus, 3R tau fibrils can convert only 3R tau, and 4R tau fibrils can convert only 4R tau 8). Thus, despite differing only by one repeat (31 amino acid), 3R tau and 4R tau behave as different proteins in seeded aggregation. These results indicate that fibrillization and conversion of normal proteins are similar to those seen in the case of Prp prion protein, which is the cause of prion diseases such as CJD and BSE.
When tau fibril seeds are directly injected into mouse brain, as is done for experimental Prp prion infection, endogenous tau proteins in the mouse brain exposed to the seeds are converted to abnormal form and phosphorylated tau pathologies develop within a few months after injection. In this case, reagents such as lipofectamine are not necessary. Most of the injected fibrils are degraded, but some are incorporated into cells, probably by endocytosis. Thus, the prion-like transmission of intracellular abnormal tau and alpha-synuclein proteins has been experimentally demonstrated by inoculation of recombinant proteins into wild-type mice.
Experimental evidence of prion-like tau propagation was first obtained in transgenic mice without tau pathology overexpressing wild-type human tau by inoculation of brain extracts of P301S mutant tau mice, which do develop tau pathology9). Subsequently, it was shown that inoculation into Tg mouse brain of brain extracts prepared from patients with various diseases, such as AD, PSP and AGD, induced tau pathologies similar to those in the human brains10). However, Tg mice could not reproduce AD-type tau pathologies composed of both 3R tau and 4R tau isoforms, because mice only express 4R tau. As already mentioned, both 3R tau and 4R tau isoforms are accumulated in AD brain in a ratio of approximately 1 : 1, so it will be necessary to generate a mouse line that expresses both 3R and 4R tau isoforms. As in the case of prion disease infection experiments, non-overexpression systems such as knock-in mice might be preferable to overexpression systems in order to recapitulate the disease conditions in the brains of patients. It will also be necessary to investigate whether the abnormal tau generated in the mice has the same biochemical and structural properties as those in the corresponding human disease.
Molecular mechanisms of tau aggregation and propagation
Lastly, I would like to discuss the molecular mechanisms of tau fibril formation and propagation in neurons of AD brain (Fig. 3). The following scenario is envisaged. Due to aging, as well as genetic and environmental factors, 3R and 4R tau interact each other and form nuclei of tau aggregation in certain neurons. Then, normal tau proteins (both 3R tau and 4R tau isoforms) interact with these seeds and assemble into tau fibrils by elongation reaction, amplifying the abnormal tau. Next, pathological tau proteins transmit to other neighboring cells and the same lesion spreads. The molecular mechanisms of cell-to-cell-transmission of intracellular proteins remain unknown, but several pathways may be possible. The protein seeds may be directly released extracellularly by cell death or some other mechanism, and incorporated into other cells by receptor-mediated or unmediated endocytosis, or through tunneling nanotubes, synapses, or unknown mechanisms. Several pathways may be involved in the transmission, but it remains to be seen which is the major one. We recently demonstrated that inoculation of a-synuclein fibrils into the striatum of marmosets (a small primate) induced Lewy body-like pathological a-synuclein aggregates in the substantia nigra at three months after inoculation. Accumulation of pathological a-synuclein and decrease of tyrosine hydroxylase (TH)-positive neurons were observed in the marmosets10). These findings strongly suggest that pathological a-synuclein is propagated retrogradely. It is difficult to explain this directional propagation of pathological proteins in terms of simple diffusion or endocytosis and exocytosis, in which secreted proteins are transmitted to the neighboring cells. The protein may propagate between cells that are connected by neuronal circuits and cellular networks. It is pharmaceutically very important to regulate the transmission of intracellular pathological proteins, such as tau in AD, because their propagation is closely correlated with disease progression. In the case of AD in particular, it should be possible to monitor the efficacy of candidate drugs or treatments to prevent tau aggregation and disease progression by means of tau PET imaging11, 12). Indeed, tau-targeted therapies using tau antibodies, which are expected to promote the removal of abnormal tau, are currently under investigation by several pharmaceutical companies, and offer new hope of overcoming AD and other neurodegenerative diseases in the future.
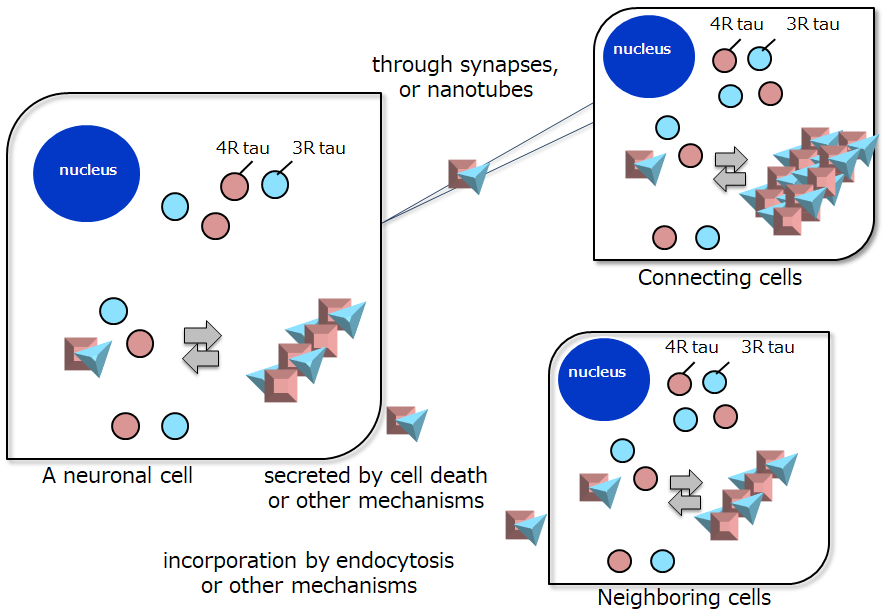
Figure 3. Schematic illustration of proposed pathways of tau aggregation and propagation in AD.
References
- Goedert M, Jakes R. Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 9: 4225-30, 1990.
- Weingarten et al. :A protein factor essential for microtubule assembly. Proc Natl Acad Sci USA. 72 : 1858-62, 1975.
- Watanabe A, et al. : In vivo phosphorylation sites in fetal and adult rat tau. J Biol Chem. 268: 25712-7, 1993.
- Hasegawa M. Molecular mechanisms in the pathogenesis of Alzheimer's disease and tauopathies - Prion-like seeded aggregation and phosphorylation. Biomolecules. 6(2), 2016.
- Braak H, Braak E. :Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. 82 : 239-59, 1991
- Goedert M. : Alzheimer's and Parkinson's diseases: The prion concept in relation to assembled Aβ, tau, and α-synuclein. Science. 349 : 1255555, 2015.
- Taniguchi-Watanabe S et al. : Biochemical classification of tauopathies by immunoblot, protein sequence and mass spectrometric analyses of sarkosyl-insoluble and trypsin-resistant tau. Acta Neuropathol. 131: 267-80, 2016.
- Nonaka T et al. :Seeded aggregation and toxicity of alpha-synuclein and tau : cellular models of neurodegenerative diseases. J Biol Chem. 285, 34885-98, 2010
- Clavaguera F, et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci U S A. 110: 9535-40, 2013.
- Shimozawa et al. Propagation of pathological α-synuclein in marmoset brain. Acta Neuropathol Commun. 5:12, 2017.
- Harada R, et al. Characteristics of tau and its ligands in PET imaging. Biomolecules 6:7, 2016.
- Ono M, et al. Distinct binding of PET ligands PBB3 and AV-1451 to tau fibril strains in neurodegenerative tauopathies. Brain 140: 764-780, 2017.