Vol. 6. α-Synuclein in Parkinson's Disease
This article was written by Dr. Norihito Uemura, Department of Neurology, Graduate School of Medicine, Kyoto University.
Introduction
Parkinson's disease (PD) is pathologically characterized by dopaminergic neuronal loss in the substantia nigra pars compacta and formation of neuronal inclusion called Lewy body. The significance of this neuronal inclusion in the pathogenesis of PD remained unknown for decades since it was first described by Fritz Heinrich Lewy in 1912.1) However, in 1997, it was reported that missense mutations in α-Synuclein (α-Syn) causing familial PD, and Lewy bodies in sporadic PD were intensely stained with the anti-α-Syn antibody, indicating a close involvement of α-Syn in the pathogenesis of PD.2,3) Given additional reports of SNCA (α-Syn gene) duplication or triplication causing familial PD and single nucleotide polymorphisms of SNCA as risk factors for sporadic PD, there is no doubt that α-Syn plays a central role in the pathophysiology of PD.4,5) This article first summarizes the physiological function of α-Syn and then reviews the involvement of α-Syn in the pathophysiology of PD.
Structure and physiological function of α-Synuclein
Synuclein was first identified in mammals in 1991, and three subtypes, that is, α-, β-, and γ-synuclein, have been identified to date.6) α-Syn, which consists of 140 amino acids, is primarily expressed in neurons in the central nervous system, especially in the presynaptic terminal. α-Syn has 7-fold repeated sequence consisting of highly conserved 6 amino acid residues in the N-terminal that is predicted to form an amphipathic α-helix structure.7) In fact, the α-Syn monomer exists in equilibrium between cytosolic, unfolded form and membrane-bound α-helical form in the cytoplasm.8) However, some researchers reported that α-Syn physiologically exists as helically folded tetramers.9) Missense mutations causing familial PD concentrate in the N-terminal region (Fig. 1). The non-amyloid-component (NAC) domain at the center of α-Syn was discovered as a peptide sequence in senile plaques in the Alzheimer's disease brains.10) This domain is hydrophobic and involved in α-Syn aggregation.11) The C-terminal is abundant in acidic amino acids and contains Ser129, which is highly phosphorylated in Lewy bodies.12) Since most α-Syn is not phosphorylated at Ser129 in the normal brains, antibodies against phosphorylated α-Syn are often used to detect pathological α-Syn aggregation.
The physiological function of α-Syn remains largely unknown. Young zebra finches learning to sing show increased expression of α-Syn in the relevant nuclei, indicating the involvement of α-Syn in neural plasticity.13) α-Syn deficient mice do not show any abnormalities in life-span, reproductive function, or brain morphology.14) However, an electrophysiological analysis of brain slice preparations revealed that α-Syn inhibited synaptic vesicle release.14) A similar function of α-Syn was also shown in a study using primary cultured neurons overexpressing α-Syn.15) In addition, α-Syn inhibits soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex-mediated lipid membrane fusion, supporting these findings.16) On the other hand, cysteine string protein-α (CSPα) promotes SNARE complex formation in the presynaptic terminal, whereas phenotypic abnormalities in CSPα-deficient mice, including synaptic degeneration and premature death, were compensated by α-Syn overexpression.17) Later, α-Syn was reported to promote SNARE complex formation by binding to SNARE synaptobrevin 2 in its C-terminal sequence.18) However, it remains unclear how to link the two functions, α-Syn-induced inhibition of synaptic vesicle release and promotion of SNARE complex formation.

Figure 1. Structure of α-Synuclein
Red: missense mutation sites reported in familial PD
Underlined: repeating sequence
Green: post-translational modification sites reported in PD
U: ubiquitination P: phosphorylation N: nitration
α-Synuclein aggregation and cytotoxicity
α-Syn is naturally a soluble protein, but forms β-sheet-rich amyloid fibrils in a group of diseases collectively called synucleinopathy. Synucleinopathy includes Lewy body dementia (DLB) and multiple system atrophy (MSA), in addition to PD. Recombinant α-Syn, which also forms fibrils in vitro, forms soluble oligomers during this aggregation process, and the oligomers and fibrils are cytotoxic (Fig. 2). Both A53T and A30P mutations cause familial PD, but the former promotes fibrillization, whereas the latter promotes oligomerization in vitro.19) Researchers insisting that α-Syn physiologically exists as tetramers reported that PD-causing missense mutations in α-Syn promote α-Syn aggregation and enhance its cytotoxicity by decreasing tetramers and increasing unfolded monomers.20) Since reactive oxygen species are produced during the metabolic process of dopamine, dopaminergic neurons are exposed to high oxidative stress. α-Syn is oligomerized through dityrosine bond under oxidative stress.21) Likewise, quinones, which are oxidative metabolites of dopamine, act on the C-terminal of α-Syn to promote and stabilize oligomerization.22) However, oligomers are not a single molecular species, but oligomers that differ in structure and cytotoxicity have been reported both in vitro and in vivo.23,24) While the oligomers may include various molecular species, the oligomers generally damage the membrane structures and cause cellular toxicity through various mechanisms, including induction of mitochondrial dysfunction and oxidative stress, impairment of endoplasmic reticulum-Golgi vesicular transport, chaperone-mediated autophagy, and SNARE complex formation in the presynaptic terminal.23-28) On the other hand, the sequence in the NAC domain is required for α-Syn aggregation,11) and solid-state nuclear magnetic resonance (SSNMR) spectroscopy revealed that 46th-54th amino acids and 63rd-96th amino acids form a Greek key topology to serves as the fibril core.29) After transduction of α-Syn fibrils into mouse primary neurons, α-Syn fibrils taken up by neurons work as templates to induce α-Syn aggregation in neurons, resulting in impairment of synaptic function and neuronal loss in 2 weeks.30) To consider the therapeutic target, it may be important to determine which (oligomers or fibrils) makes a greater contribution to the pathogenesis of PD. Oligomers are more toxic than fibrils in vivo,31) and oligomers unidentifiable with usual immunostaining for α-Syn are extensively detected with the proximity ligation assay (PLA) in the brain of patients with PD.32) On the other hand, an in vivo imaging analysis revealed aggregate-bearing neurons die over time in α-Syn fibril-injected mouse model.33) It still remains controversial which (oligomers or fibrils) makes a greater contribution to the pathogenesis of PD.
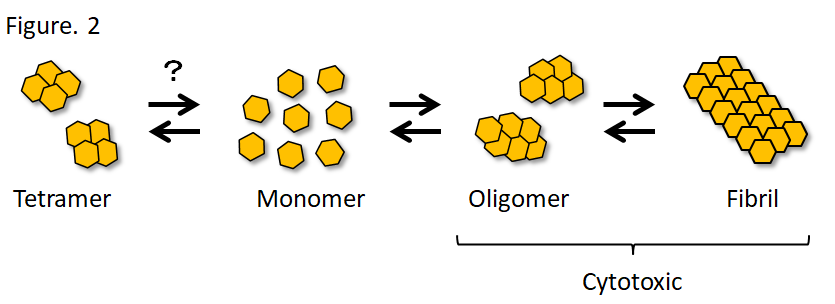
Figure 2. α-Synuclein aggregation
It is still controversial whether α-Syn physiologically exists as monomers or tetramers. It is suggested that there are distinct oligomers and fibrils that differ in structure and biological activity.
Intercellular spreading of α-Synuclein
Lewy pathology is not limited to dopaminergic neurons in the substantia nigra pars compacta but is widely seen in other brain regions. Braak et al. systematically analyzed the pathology in cases with incidental Lewy body pathology or sporadic PD and suggested that Lewy pathology initially develops in the olfactory bulb and the dorsal motor nucleus of the vagus nerve and then spreads in the brain stereotypically.34) In addition, since Lewy pathology is also found in the enteric nervous system (ENS) in the early stage of PD, they hypothesized that Lewy pathology in the ENS travels retrogradely to the dorsal motor nucleus of the vagus nerve and then proceeds from there in a caudo-rostral direction.35) Despite the impact of the Braak's hypothesis on investigation of the clinicopathologic progression of PD, this hypothesis lacked the evidence that Lewy pathology can spread among brain regions. However, the patients with PD who underwent intracerebral transplantation of fetal mesencephalic dopaminergic neurons developed Lewy bodies in the graft cells, suggesting that Lewy pathology can propagate from host to graft cells.36,37) In addition, it was shown that injection of α-Syn fibrils formed in vitro into the mouse brain induces aggregation of endogenous α-Syn in neurons, which then spread along the neural circuit.38,39) Injection of Lewy bodies extracted from the PD brains into the mouse or monkey brains also induces α-Syn aggregate formation in host neurons, which then spread to other brain regions.40) This phenomenon is known as prion-like spreading of α-Syn and may explain the Braak hypothesis, attracting wide interest of researchers. From the aspect of therapeutic target, researchers are trying to elucidate the mechanisms of interneuronal spreading of α-Syn and find how to inhibit the spreading. While various possible mechanisms of the spreading, including direct cell membrane penetration, macropinocytosis, receptor-mediated endocytosis, and exosome- or nanotube-mediated transfer, have been reported, it is still unclear which mechanism plays the most important role in the pathogenesis of PD.41-45)
Structural polymorphism in α-Synuclein aggregates
It has recently been reported that α-Syn fibrils formed in vitro differ in biochemical properties (aggregate structure) and pathology induced in cultured cells or mouse brains (biological activity) depending on conditions in which α-Syn fibrils are formed,46,47) indicating the existence of diverse α-Syn aggregates, more specifically, the possibility that distinct α-Syn aggregates may induce different pathologies in humans. PD and DLB are both characterized by α-Syn aggregates in neurons but differ in clinical course and pattern of pathological progression. MSA is characterized by α-Syn aggregates in oligodendrocytes and clearly differs from PD and DLB. To date, the mechanisms by which the same pathological protein, α-Syn, causes distinct synucleinopathies remain one of the unsolved mysteries in this field. Several recent studies have demonstrated the differences of pathological α-Syn aggregates in distinct synucleinopathies. For instance, pathological α-Syn extracted from MSA brains is more potent for inducing α-Syn aggregation and more toxic in the mouse brains or cultured cells than one extracted from PD brains.48,49) However, the pathological differences in distinct synucleinopathies cannot be explained only by the differences of pathological α-Syn aggregates because injection of pathological α-Syn extracted from MSA brains into the mouse brains result in α-Syn aggregates in neurons, but not in oligodendrocytes.48,50) In addition to the differences in α-Syn aggregates, some host factors may be necessary to explain and reproduce the pathological differences in distinct synucleinopathies.
Conclusion
While approximately 20 years have passed since identification of α-Syn as the main component of Lewy body, various pathological mechanisms and therapeutic targets have been reported, but have not led to the development of disease-modifying therapy. Because no experimental system can reproduce the disease pathology completely, it should be important to establish a system which reproduces well a certain pathological mechanism and try to develop therapeutic intervention against it.
References
- Goedert, M. et al . : Nat. Rev. Neurol ., 9, 13(2013)
- Polymeropoulos, M. H. et al . : Science , 276,2045(1997)
- Spillantini, M. G. et al . : Nature , 388, 839(1997)
- Singleton, A. B. et al . : Science , 302, 841(2003)
- Satake, W. et al . : Nat. Genet ., 41, 1303(2009)
- Maroteaux, L. and Scheller, R. H. : Brain Res . Mol. Brain Res. , 11, 335(1991)
- Bendor, J. T. et al . : Neuron , 79, 1044(2013)
- Pineda, A. and Burre, J. : Proc. Natl. Acad. Sci. USA, 114, 1223(2017)
- Bartels, T. et al . : Nature , 477, 107(2011)
- Ueda, K. et al . : Proc. Natl. Acad. Sci. USA , 90, 11282(1993)
- Giasson, B. I. et al . : J . Biol. Chem ., 276, 2380(2001)
- Fujiwara, H. et al . : Nat. Cell Biol ., 4, 160(2002)
- George, J. M. et al . : Neuron , 15, 361(1995)
- Abeliovich, A. et al . : Neuron , 25, 239(2000)
- Nemani, V. M. et al . : Neuron , 65, 66(2010)
- DeWitt, D. C. and Rhoades, E. : Biochemistry ,52, 2385(2013)
- Chandra, S. et al . : Cell , 123, 383(2005)
- Burre, J. et al . : Science , 329, 1663(2010)
- Conway, K. A. et al . : Proc. Natl. Acad. Sci. USA, 97, 571(2000)
- Dettmer, U. et al . : Nat. Commun ., 6, 7314(2015)
- Souza, J. M. et al . : J. Bio. Chem ., 275, 18344(2000)
- Mazzulli, J. R. et al . : J. Neurosci ., 26, 10068(2006)
- Fusco, G. et al . : Science , 358, 1440(2017)
- Mor, D. E. et al . : Nat. Neurosci ., 20, 1560(2017)
- Mazzulli, J. R. et al . : Cell , 146, 37(2011)
- Cremades, N. et al . : Cell , 149, 1048(2012)
- Martinez-Vicente, M. et al . : J. Clin. Invest . 118, 777(2008)
- Choi, B. K. et al . : Proc. Natl. Acad. Sci. USA, 110, 4087(2013)
- Tuttle, M. D. et al . : Nat. Struct. Mol. Biol .,23, 409(2016)
- Volpicelli-Daley, L. A. et al . : Neuron , 72, 57(2011)
- Winner, B. et al . : Proc. Natl. Acad. Sci. USA, 108, 4194(2011)
- Roberts, R. F. et al . : Brain , 138, 1642(2015)
- Osterberg, V. R. et al . : Cell Rep ., 10, 1252(2015)
- Braak, H. et al . : Neurobiol. Aging , 24, 197(2003)
- Braak, H. et al . : Neurosci. Lett ., 396, 67(2006)
- Li, J. Y. et al . : Nat. Med ., 14, 501(2008)
- Kordower, J. H. et al . : Nat. Med ., 14, 504(2008)
- Luk, K. C. et al . : Science , 338, 949(2012)
- Masuda-Suzukake, M. et al . : Brain , 136, 1128(2013)
- Recasens, A. et al . : Ann. Neurol ., 75, 351(2014)
- Lee, H. J. et al . : Int. J. Biochem. Cell Biol .,40, 1835(2008)
- Hansen, C. et al . : J. Clin. Invest ., 121, 715(2011)
- Alvarez-Erviti, L. et al . : Neurobiol. Dis ., 42, 360(2011)
- Holmes, B. B. et al . : Proc. Natl. Acad. Sci. USA, 110, E3138(2013)
- Abounit, S. et al . : EMBO J ., 35, 2120(2016)
- Guo, J. L. et al . : Cell , 154, 103(2013)
- Peelaerts, W. et al . : Nature , 522, 340(2015)
- Prusiner, S. B. et al . : Proc. Natl. Acad. Sci. USA, 112, E5308(2015)
- Woerman, A. L. et al . : Proc. Natl. Acad. Sci. USA, 112, E4949(2015)
- Bernis, M. E. et al . : Acta Neuropathol. Commun ., 3, 75(2015)