Regulation of Plasticity through the BDNF Pathway and its Role in Psychiatric Disorders
This article was written by Juzoh Umemori, PhD, Docent, A.I. Virtanen Institute, University of Eastern Finland.
Introduction
The Brain-derived neurotrophic factor (BDNF) regulates neural plasticity, such as neuronal survival and differentiation, by acting on its receptor Neurotrophic receptor tyrosine kinase 2 (Ntrk2, TrkB). As neural plasticity increases, neural circuits are remodeled both structurally and functionally, adapting to environmental changes or promoting learning and memory. Pathophysiological (pre)clinical studies have reported impaired signaling through BDNF /TrkB pathway in psychiatric disorders such as schizophrenia and depression. Recently, the molecular and network mechanisms by which antidepressants modulate neural plasticity through BDNF/TrkB signaling have also been elucidated. In this article, we will discuss the role of an impaired BDNF/TrkB pathway in psychiatric disorders, and how targeting this pathway can ameliorate neuropsychiatric-like symptoms.
Impaired BDNF/TrkB Pathway in Schizophrenia
The BDNF/TrkB pathway plays a crucial role in neuronal development during the embryonic and critical periods, as well as in the regulation of neuronal plasticity and survival in the hippocampus and cortex in adults. Animal models of schizophrenia show BDNF dysregulation, and forebrain-specific TrkB knockout mice exhibit schizophrenia-like behaviors such as hyperkinesis, stereotypic behaviors, and cognitive deficits. In addition, studies of postmortem brains from schizophrenic subjects revealed decreased BDNF concentrations in cortical regions and the hippocampus, whereas other authors reported increased BDNF concentrations in the anterior cingulate cortex and hippocampus. It has been suggested that these aberrant BDNF expression patterns may result in abnormal neurodevelopment and decreased neuroplasticity in schizophrenic patients 1-3).
BDNF/TrkB Signaling Abnormalities in Mood Disorders
BDNF messenger RNA and protein expression are decreased in the postmortem brains of suicide victims and patients with major depressive disorder, particularly in the hippocampus and amygdala 4). The DNA methylation of the BDNF gene promoter is increased in peripheral blood mononuclear cells of suicide victims and patients with depression, suggesting a dysregulation of BDNF expression 5). High levels of BDNF, presumably derived from platelets, can be detected in human serum. Interestingly, patients with depression exhibit lower BDNF levels in the serum or plasma compared to successfully treated patients and healthy controls 4, 6). The serum BDNF/proBDNF ratio is higher in patients with bipolar disorder and lower in patients with depression compared to healthy controls and is therefore proposed as a potential marker that can be used to distinguish between bipolar and major depressive disorder 7, 8). In contrast, as noted earlier, BDNF levels are decreased in patients with schizophrenia 1, 3), while both elevated and decreased BDNF levels have been reported in patients with autism 9). Serum BDNF levels can exhibit significant inter- and intra-individual variability, and further studies are needed to determine the reliability and disease-specificity of serum BDNF as a marker of disease. In addition to BDNF, TrkB mRNA and protein levels are also decreased in the postmortem brains of patients with depression, and genetic mutations in TrkB have been associated with suicide behavior. Furthermore, the active, phosphorylated form of TrkB is decreased in brain samples of patients with depression 4), suggesting an additional connection between the BDNF pathway function and modulation of neuroplasticity.
Genetic Association Between BDNF and Psychiatric Disorders
25% to 50% of Caucasian populations carry an SNP variation in the BNDF sequence that causes substitution of the predominant valine to methionine (Val66Met polymorphism) 10, 11). This polymorphism is suggested to be a risk factor for psychiatric disorders such as depression and schizophrenia, but also for neurodegenerative diseases including multiple sclerosis, Alzheimer's disease, and Parkinson's disease 12, 13). A study reported that this polymorphism is also involved in the susceptibility to depression caused by childhood abuse, poverty, and chronic stress in adulthood 14). In animal studies, this polymorphism has been shown to affect spatial memory, anxiety-like behavior, intracellular BDNF transport and neuronal activity-dependent BDNF release 10, 15), while also impairing dendritic transport of BDNF mRNA 16), and reducing long-term depression (LTD) 17). Taken together, these results suggest that the Val66Met polymorphism may affect the BDNF-dependent modulation of neuroplasticity.
Increased Plasticity Caused by Antidepressants Through BDNF/TrkB Signaling
Given that BDNF/TrkB signaling is decreased in the blood and brain of patients with depression, the finding that antidepressants can restore BDNF levels is auspicious. In fact, animal studies have shown that tricyclic monoamine oxidase inhibitors and serotonin-selective reuptake inhibitors (SSRIs) increase BDNF expression in the brain 6). The effects of antidepressants on depressive-like behavior and neurogenesis are reduced by the deletion of BDNF from mouse hippocampal dentate gyrus cells and by the deletion of TrkB expression from progenitor cells of dentate gyrus granule neurons 6). In addition, the fast-acting antidepressant effects of the anesthetic ketamine (an antagonist of NMDA-type glutamate receptors) such as reduction of depressive-like behavior and hippocampal synaptic enhancement, are also suppressed by the deletion of the BDNF or TrkB gene in the mouse forebrain region 18). Interestingly, behavioral and neuroplasticity-related responses to antidepressants are also lost in mouse models of the Val66Met polymorphism 15, 19). Furthermore, the increase in spine formation induced by ketamine and its metabolite, (2R, 6R)-hydroxynorketamine (HNK), primarily responsible for the antidepressant effects of ketamine via BDNF/TrkB signaling 21), was suppressed in these mice 18, 20).
Some clinical studies, however, suggest that the Met allele improves response to conventional antidepressants 22-25), whereas others have reported a decrease in response 26). Additional genetic analyses are therefore needed to elucidate the role of BDNF signaling in antidepressant response in patients with depression 4).
Improvement of Neuropsychiatric-like Symptoms by Induced Juvenile-like Plasticity (iPlasticity)
Research from the Neurotrophin Laboratory at the University of Helsinki has shown that chronic administration of antidepressants induces a juvenile‐like or cirital period-like plasticity. Prof. Eero Castren proposes that the promoted plasticity, combined with some type of "training," could be applied to the treatment of depression as well as other neuropsychiatric disorders 27). For instance, chronic administration of fluoxetine, a common SSRI, to rats or mice, followed by monocular deprivation improves symptoms in models of amblyopia, while in combination with fear memory extinction training alleviate post-traumatic stress disorder (PTSD), and when combined with social skills training leads to reduced aggression 28-31). While the fluoxetine-mediated increase in neuroplasticity was thought to occur through BDNF/TrkB activation and enhanced remodeling of the neural network upon neural activation by internal and external stimulation, how antidepressants activate TrkB and which neural networks underwent what type of reorganization remained unknown. A recent study has demonstrated that antidepressants belonging to various classes including fluoxetine, imipramine, and ketamine can directly bind TrkB 32). TrkB forms a dimer featuring a cross-like conformation of monomers in the transmembrane domain, creating a pocket in which antidepressants may bind. Antidepressants bind directly to the cholesterol-binding domain in this pocket, stabilizing TrkB in the synaptic membrane and enhancing BDNF/TrkB signaling 32). These findings have led to an entirely new hypothesis by which the primary site of action of antidepressants occurs via direct binding to TrkB rather than to monoamine transporters, as was generally believed, generating a great discussion. Our current work utilizes optically activatable TrkB (optoTrkB), which dimerizes upon photoactivation, to examine the plasticity of target cells, a process which has been difficult to study given the diffusible nature of BDNF. We have demonstrated that TrkB activation in parvalbumin-positive interneurons induces a shift of the ocular dominance when combined with monocular deprivation, promotes synaptic plasticity, and potentiates LTP and synchronized EEG oscillations caused by disinhibition 33). Working with the optoTrkB system, we aim to develop/aid the development of PTSD treatment, improve spatial memory, and reduce drug addiction.
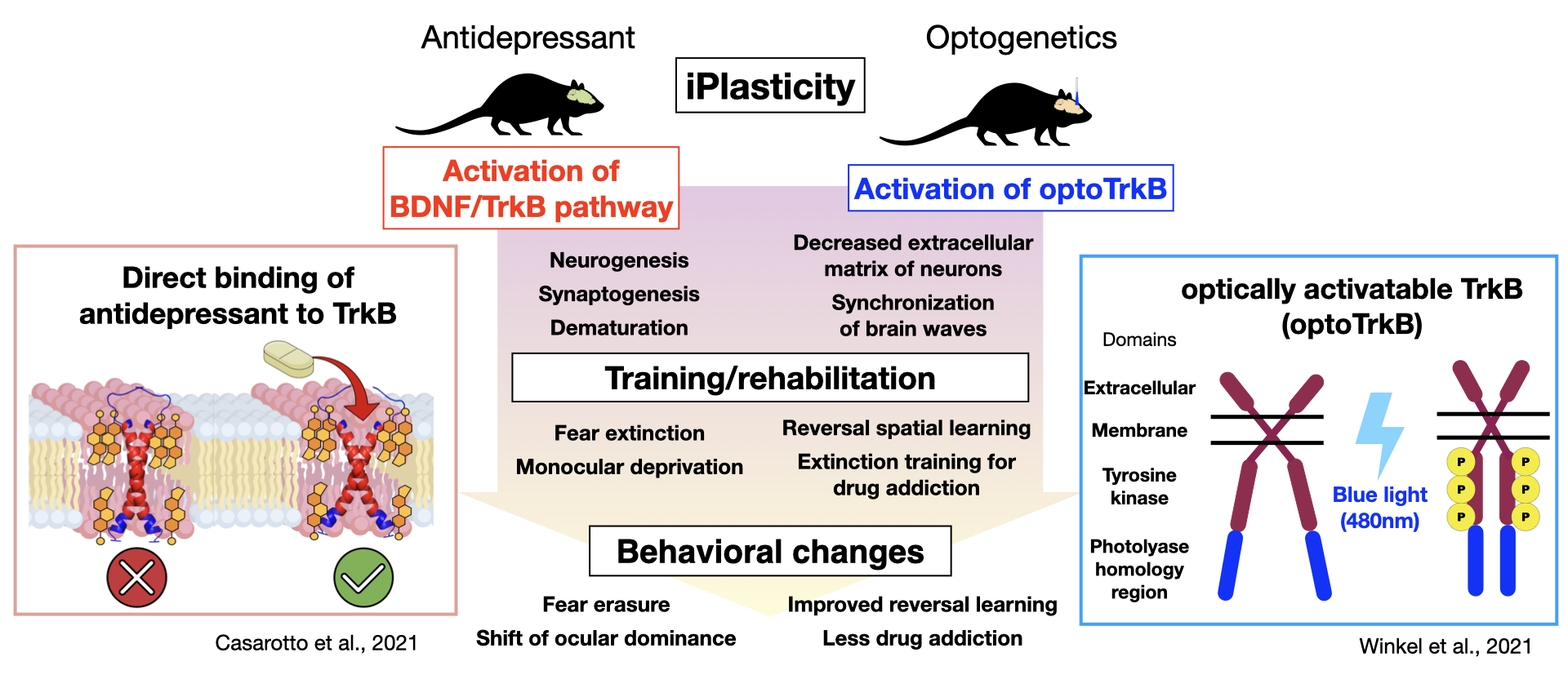
Conclusion
Since the discovery of the Val66Met polymorphism, numerous genetic studies have examined its role in the different responses to antidepressants. Failure to replicate these effects in some studies may be due to complex interactions among various factors, including age, gender, ethnicity, and environmental factors, as well as the genetic model used in the study and any genetic interactions 34). The Val66Met polymorphism mouse model and the new optoTrkB model of antidepressant action are expected to help elucidate the pathogenic mechanisms of psychiatric disorders involving the BDNF/TrkB pathway to find new treatment options.
Acknowledgments
I would like to give thanks to Dr. Plinio Casarotto of Elsevier for the picture, Dr. Koshimizu of Fujita Health University and Mr. Giuliano Didio of University of Helsinki for their helpful advice.
Reference
- Angelucci, F. et al. : Mol. Psychiatry, 10 (4), 345 (2005).
- Fernandes, B. S. et al. : Mol. Psychiatry, 20, 1108 (2015).
- Nieto, R. R. et al. : Front. Psychiatry, 12, 662407 (2021).
- Castrén, E. and Monteggia, L. M. : Biol. Psychiatry, 90, 128 (2021).
- Hing, B. et al. : Am. J. Med. Genet. B Neuropsychiatr. Genet., 177, 143 (2018).
- Autry, A. E. and Monteggia, L. M. : Pharmacol. Rev., 64, 238 (2012).
- Yoshida, T. et al. : PLoS One, 7, e42676 (2012).
- Södersten, K. et al. : J. Affect. Disord., 160, 1 (2014).
- Saghazadeh, A. and Rezaei, N. : J. Autism Dev. Disord., 47, 1018 (2017).
- Egan, M. F. et al. : Cell, 112, 257 (2003).
- Shimizu, E. et al. : Am. J. Med. Genet. B Neuropsychiatr. Genet., 126B, 122 (2004).
- Bath, K. G. and Lee, F. S. : Cogn. Affect. Behav. Neurosci., 6, 79 (2006).
- Shen, T. et al. : Aging Dis., 9, 523 (2018).
- Hosang, G. M. et al. : BMC Med., 12 : 7, 1 (2014).
- Chen, Z.-Y. et al. : Science, 314, 140 (2006).
- Baj, G. et al. : Front. Neurosci., 7, 188 (2013).
- Mizui, T. et al. : Proc. Natl. Acad. Sci. U. S. A., 112, E3067 (2015).
- Björkholm, C. and Monteggia, L. M. : Neuropharmacology, 102, 72 (2016).
- Bath, K. G. et al. : Neuropsychopharmacology, 37, 1297 (2012).
- Fukumoto, K. et al. : Proc. Natl. Acad. Sci. U. S. A., 116, 297 (2019).
- Zanos, P. et al. : Nature, 533, 481 (2016).
- Choi, M. J. et al. : Brain Res., 1118, 176 (2006).
- Niitsu, T. et al. : Prog. Neuro-psychopharmacol. Biol. Psychiatry, 45, 183 (2013).
- Yan, T. et al. : Asia-Pac. Psychiatry, 6, 241 (2014).
- Domschke, K. et al. : Int. J. Neuropsychopharmacol., 13, 93 (2010).
- Laje, G. et al. : Biol. Psychiatry, 72, e27 (2012).
- Castrén, E. : Nat. Rev. Neurosci., 6, 241 (2005).
- Umemori, J. et al. : Psychiatry Clin. Neurosci., 72, 633 (2018).
- Mikics, É. et al. : Neuropsychopharmacology, 43, 235 (2017).
- Karpova, N. N. et al. : Science, 334, 1731 (2011).
- Vetencourt, J. F. M. et al. : Science, 320, 385 (2008).
- Casarotto, P. C. et al. : Cell(2021). doi:10.1016/j.cell.2021.01.034.
- Winkel, F. et al. : Mol. Psychiatry, 1-10 (2021). doi : 10.1038/s41380-021-01211-0.
- Tsai, S.-J. et al. : Front. Mol. Neurosci., 11, 156 (2018).