Another face of BDNF
This article was written by Naoki Adachi, Department of Physiology, School of Medicine, Showa University.
Introduction
BDNF/TrkB signaling pathways play beneficial functions in the normal development of neural circuits and regulation of neurotransmission. Therefore, disruption of BDNF/TrkB signaling is a cause of diseases in the central nervous system (CNS) and other tissues, and many drug discovery studies have been conducted to target its activation. However, in this article, I would like to discuss several cases in which activation of BDNF/TrkB signalings can be an "enemy" to our health. First, I will review the main BDNF/TrkB signaling pathways and introduce the involvement of BDNF/TrkB signaling activation in individual diseases.
BDNF/TrkB signaling pathways
1. MAPK/ERK pathway
When the Tyr490 and Tyr515 residues of TrkB receptor are activated, the adapter protein Shc docks onto these tyrosine sites and recruits Grb2 (growth factor receptor binding protein 2) and Ras MAPK/ERK kinase, which in turn phosphorylates a transcription factor CREB (cAMP response element binding protein). Phosphorylated CREB is translocated into the nucleus and binds to promoters of various genes, thereby regulating transcription to promote cell survival, differentiation, and proliferation (Figure 1).
2. PI3K/Akt pathway
The Ras complex with Tyr515 of TrkB also activates the PI3 (phosphoinositide 3) kinase pathway that activates two cascades: the PI3K/Akt pathway and the MEK/MAPK pathway. The PI3K/Akt pathway also regulates a group of proteins essential for differentiation, growth, and survival (Figure 1).
3. PLCγ pathway
Phosphorylation of Tyr816 residue of TrkB receptor activates the PLCγ (phospholipase Cγ) pathway, producing DAG (diacylglycerol) and IP3 (inositol-1,4,5-triphosphate). The PLCγ/IP3 pathway stimulates Ca2+ releases from the ER and activates CaMKII (calcium/calmodulin dependent protein kinase II) that induces CREB phosphorylation while DAG activates PKC (protein kinase C). Activation of CaMKII and PKC has roles to promote neurite outgrowth, synaptic plasticity, and survival of neurons (Figure 1).
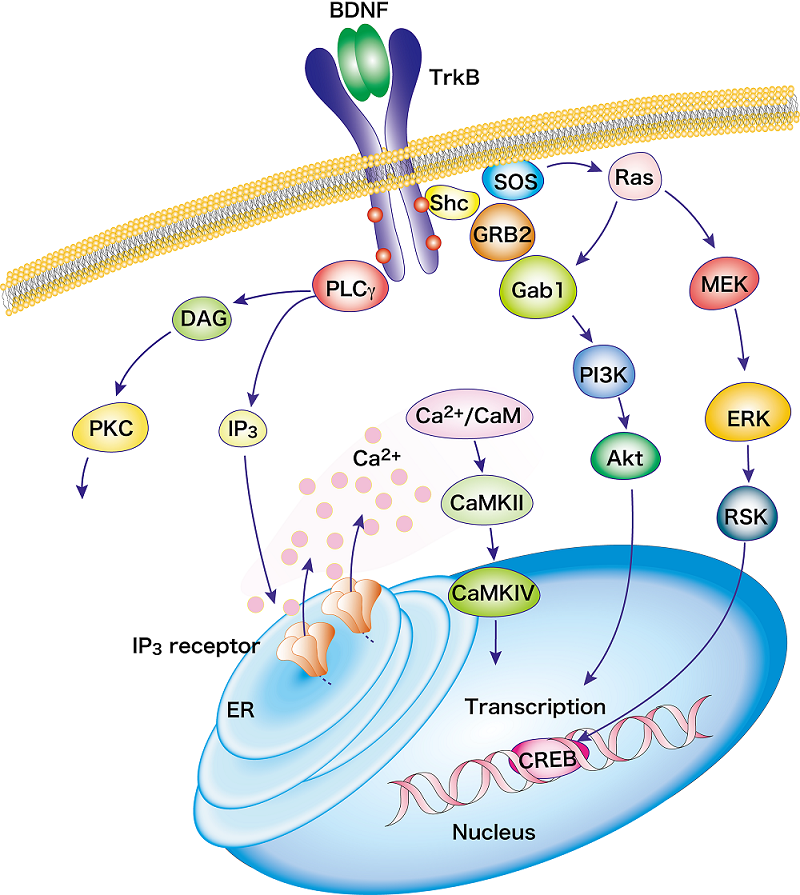 Figure 1. BDNF/TrkB signaling pathway Modified from the authors' reference38)
Figure 1. BDNF/TrkB signaling pathway Modified from the authors' reference38)
BDNF/TrkB Signaling in Drug Addiction
BDNF/TrkB signaling plays several roles in drug addiction, including cocaine addiction, by promoting plastic changes of neural circuits in the brain's "reward system" 1). Among the brain regions responsible for the reward system, high level expression of BDNF is observed in the amygdala, ventral tegmental area (VTA), hippocampus, and frontal cortex. On the other hand, its expression is low in the nucleus accumbens (NAc) and dorsal striatum2). In contrast to BDNF, TrkB expression is widely observed in many brain regions3). In rodent studies, cocaine exposure increased BDNF expression levels in the reward-related regions4,5). In the studies using cocaine addiction animal models, BDNF infusion into the VTA or NAc, or forced expression of BDNF/TrkB enhanced conditioning place preference, cocaine-seeking behavior, and withdrawal symptoms, while infusion of anti-BDNF antibodies or knockdown of BDNF/TrkB inhibits these reward-related behaviors6). A model has been proposed in which cocaine-induced enhancement of BDNF/TrkB signaling in the NAc and other regions is responsible for the development of addiction symptoms by modulating the organization of AMPA glutamate receptor subunits at the transcriptional and translational levels, causing plastic synaptic changes1). On the other hand, since BDNF/TrkB signaling enhancement in the medial prefrontal cortex (mPFC) plays an inhibitory role in cocaine addiction symptoms in rodent studies7), treatment targeting BDNF/TrkB signaling inhibition may be sufficiently effective, if limited to the NAc and VTA regions8).
BDNF/TrkB signaling in chronic pain
Pain sensory stimuli are transmitted by dorsal root ganglion neurons that act as primary afferents and send excitatory inputs to secondary neurons in the dorsal horn (DH) of the spinal cord. The enhanced excitability of these secondary neurons in the DH is known to be a major factor in neuropathic pain. Enhanced activation of BDNF/TrkB signaling in the DH may be significantly involved in the onset and progression of neuropathic pain through the following three suggested mechanisms.
(i) KCC2
Two distinct inhibitory mechanisms to suppress the sensation of pain exist in the spinal cord: presynaptic inhibition on primary afferents and postsynaptic inhibition on DH secondary neurons, and BDNF has been suggested to be involved in the regulation of these GABAergic (inhibitory) synapses to cause hyperalgesia9). A key protein that regulates excitability of GABAergic neurons is potassium chloride cotransporter 2 (KCC2), a transmembrane protein that ensures GABAergic inhibitory properties by pumping intracellular chloride ions (Cl-) out of the cell, thereby increasing extracellular Cl- concentration and hyperpolarizing GABAergic postsynaptic cells during GABA receptor opening10). In animal models of neuropathic pain, BDNF expression is increased and KCC2 protein expression is decreased in the DH of the spinal cord11). Furthermore, intrathecal administration or a TrkB inhibitor K252a of TrkB/Fc chimeras blocked the decrease in KCC2 expression in the spinal DH caused by peripheral nerve injury and alleviated neuropathic pain12). Although the details are still unclear, it appears that transcriptional repression of KCC2 occurs when the Shc and PLCγ pathways are activated in tandem in the downstream of BDNF/TrkB signaling activation, which reduces the inhibitory function of GABAergic neurotransmission 13).
(ii) Enhanced excitability
In models of nerve injury, activation of TrkB receptor by BDNF has been shown to promote synaptic transmission by increasing the accumulation of glutamatergic synaptic vesicles in the presynaptic area and enhancing NMDA receptor function, which may lead to the development of neuropathic pain14). BDNF is also known to increase the number of TrkB receptors in the posterior spinal cord. Binding of BDNF to TrkB receptor expressed on the surface of DH neurons in the spinal cord causes long-term potentiation of the dorsal root ganglion neuron-spinal DH neuron synapse via phosphorylation of the GluN2B (NR2B) subunit by Fyn kinase, resulting in central sensitization and neuropathic pain15,16).
(iii) Activation of glial cells
We have seen above how increased excitability of spinal DH neurons induced by BDNF causes neuropathic pain, but which cells are the first to supply BDNF in the event of nerve injury? Previous studies have shown that the source of BDNF is glial cells.
Microglia activated by peripheral nerve injury and other factors produce and release cytokines, such as, BDNF, TNF-α, and interleukins, which induces neuroimmune responses. Such a neuroinflammatory response can lead to the pathogenesis of pain sensitivity17). Activated microglial cells increase cell migration with enhanced production/release of BDNF and TNF-α18). Furthermore, BDNF activates microglia itself, suggesting a possibility of a positive circulatory loop mechanism in which microglia-derived BDNF acts as autocrine factor to further activate itself (microglia) during nerve injury19).
Astrocyte activation in the DH of the spinal cord also increases BDNF and inflammatory cytokine levels20), and astrocytes are also considered as a major component of sustained central sensitization21). Although increased BDNF synthesis/release as a response to nerve injury should be adaptive for the human body since BDNF protects neurons and promotes their survival, the absence of a negative feedback mechanism may transform the nervous system to a nonadaptive state as neuropathic pain progresses.
BDNF/TrkB Signaling in Epilepsy
Epilepsy is a progressive CNS disorder in which a group of neurons in the brain suddenly over-fire, and currently affects approximately 0.5% to 1% of the world's population22). Patients with epilepsy have recurrent spontaneous seizures, as well as serious symptoms such as brain dysfunction and cognitive decline23). In clinical studies, the majority of patients with epilepsy are known to have higher serum BDNF levels than healthy subjects. In animal models of epilepsy, BDNF and TrkB expression is increased, especially in the temporal and hippocampal regions24). In such mouse epilepsy models, deletion of TrkB25) or transient inhibition of TrkB receptors26) can significantly reduce the occurrence of epileptic seizures. Furthermore, administration of BDNF to the hippocampal region of rats can induce epileptic seizures27).
How is BDNF/TrkB signaling involved in epileptogenesis? To date, three main mechanisms have been proposed: 1) BDNF induces Ca2+ release and reactive oxygen species (ROS) production via ryanodine receptors28), which promotes neuronal damage and apoptosis29). 2) Increased TrkB activation suppresses the expression of KCC2 and the inhibitory effects of GABAergic neurons27). 3) Increased BDNF/TrkB signaling causes abnormal sprouting of mossy fibers, the axons of hippocampal dentate gyrus granule cells, making neural circuit abnormally excitated30).
Although the exact role of BDNF/TrkB in epileptogenesis remains unclear, including the amount of BDNF expressed, the brain regions where it is expressed, and its upstream and downstream molecules, proteins and functional molecules involved in BDNF/TrkB signaling are expected to serve as targets and indicators for clinical treatment, risk assessment, and prognosis prediction.
BDNF/TrkB signaling in cancer
BDNF/TrkB signaling has been shown to promote cellular oncogenesis, invasion and metastasis, and resistance to chemotherapy. When a neural crest-derived cell line overexpressing TrkB was transplanted into mice, tumors formed rapidly after 10 days, and all mice died within 7 days after tumor formation31). In addition, brain tumor-forming cells overexpress TrkB and can survive without native growth factors such as EGF as long as they can produce BDNF, suggesting that tumor cells have acquired the ability to maintain their own survival by autocrine BDNF/TrkB system32).
One factor that makes tumor treatment difficult is metastasis to other organs. From the tumor cell's point of view, metastasis is an event with risk of its own survival. It must break through the barrier of cell death (anoikis) caused by separation from the extracellular matrix33) and change its nature from epithelial to mesenchymal (epithelial-mesenchymal transition: EMT) by altering gene expression so that it can survive to metastasize via blood to distant organs33,34). Interestingly, tumor cells appear to increase their resistance to anoikis and increase the success rate of metastasis by activating PI3 kinase constitutively through TrkB activation32,33). Although the detailed mechanism is beyond the scope of this article, it is believed that BDNF/TrkB signaling acts in an inhibitory manner against endogenous and exogenous apoptosis, conferring resistance to anticancer chemotherapy35).
While BDNF/TrkB signaling contributes to cancer formation, metastasis, and resistance to therapy, "beneficial" functions have also been reported. Mice implanted with melanoma were found to have enhanced cytotoxicity by natural killer cells and T cells and significantly suppressed tumor growth under conditions of environmental enrichment36,37). It is known that BDNF expression in the hypothalamus is increased in environmentally enriched mice, and knockdown of BDNF abolishes tumor resistance induced by environmental enrichment36), suggesting that elevated BDNF in the hypothalamus is the key to the anti-tumor effect of environmental enrichment.
Conclusion
In this article, we have looked at the "another face" of BDNF/TrkB signaling, which is not conducive to our health. However, the basic properties/functions of BDNF/TrkB signaling, in which BDNF bound to TrkB activates various intracellular signaling cascades, thereby regulating synaptic transmission and promoting cell survival and proliferation, remains the same as for its beneficial functions. Whether BDNF/TrkB signaling is ectopically or excessively activated is the key to whether BDNF/TrkB signaling is a threat to our health.
In the expectation of therapeutic effects on brain diseases and dysfunction, as well as anticancer effects via the hypothalamus, allosteric modulators (collectively referred to as ampakines) that induces enhancement of the response of AMPA glutamate receptors and resultant increase of BDNF expression in the brain has been developed. One ampakine has been shown to increase BDNF expression levels up to 20-fold in several brain regions within several days of administration, but there are high barriers to the clinical application of these agents, including the appearance of unintended seizure-inducing effects. Recently, however, a bright light has emerged with the report of the first ampakine that improves age-related memory impairment without inducing side effects of seizure37).
By focusing on the negative aspects of BDNF/TrkB signaling, I hope that readers have reaffirmed the need for BDNF/TrkB signaling to be moderately activated in appropriate areas and the importance of accumulating basic research evidence for clinical application.
References
- Li, X. and Wolf, M. E. : Brain Res., 279, 240 (2015).
- Conner, J.M., et al. : Neurosci., 17, 2295 (1997).
- Krause, S., et al. : Neurosci. Res., 86, 411 (2008).
- Graham, D. L., et al. : Neurosci., 10, 1029 (2007).
- Horger, B. A., et al. : J. Neurosci., 19, 4110(1999).
- Bahi, A. et al. : Psychopharmacology (Berl), 199, 169 (2008).
- Berglind W. J. et al. : J. Neurosci., 26, 757 (2007).
- Li, X and Wolf, M. E.: Brain Res., 279, 240(2015).
- Malan T. P. et al. : Anesthesiology., 96, 1161 (2002).
- Janssen, S. P., et al. : Neurochem. Int., 60, 21 (2012).
- Malcangio, M. : Pain, 1, 1 (2017).
- Yajima, Y, et al. : J Neurochem., 93, 584 (2005).
- Lee-Hotta. S., et al. : Int., 128, 32 (2019).
- Wang, L. N. et al. : Neurosci. Res., 90, 1249 (2012).
- Li, S. et al. : Res., 42, 2712 (2017).
- Ding, X. et al. : Dis., 73, 428 (2015).
- Tsuda, M. et al. : Trends Neurosci., 28, 101 (2005).
- Zhang, X. et al. : Physiol. Biochem., 34, 715 (2014).
- W. et al. : Neurosci. Lett., 756, 135965 (2021).
- Chiang, C. Y. B.J. et al. : Res., 37, 2419 (2012).
- Liu, T. et al. : Bull., 28, 131 (2012).
- Fisher, R. S. et al. : Epilepsia, 55 , 475 (2014).
- Thijs, R. D. et al. : Lancet, 393, 689 (2019).
- Wang, X. and Z, Hu. : Pharmacol., 12, 758232 (2021).
- He, X. P. et al. : Neuron, 43, 31 (2004).
- Liu, G., et al. : Neuron, 79, 31 (2013).
- Scharfman, H. E. et al. : Neurol., 174, 201 (2002).
- Yang, N. et al. : Med. Cell Longev., 2020, 6687185 (2020).
- Ghadiri, T. et al. : Lett., 709, 134384 (2019).
- Skupien-Jaroszek, A. et al. : PLoS One, 16, e0239111.(2021).
- Yin, B. et al. : oncogene, 34, 761(2015).
- Lawn, S. et al. : Biol. Chem., 290, 3814 (2015).
- Bao, W., et al. : PLos One, 8, e70616 (2013).
- Kupferman, M. E. et al. : oncogene, 29, 2047(2010).
- Radin, D. P. : Anticancer Res., 37, 39832017 (2017).
- Cao, L. et al. : Cell, 142, 52 (2010).
- Radin, D. P. et al. : pharmacother., 84, 806 (2016).
- Numakawa, T. et al. : World J. Biol. Chem., 26, 133 (2010).