Vol.3 Amyloid β in Alzheimer's Disease
This article was written by Dr. Yukiko Hori and Dr. Taisuke Tomita, Laboratory of Neuropathology and Neuroscience, Graduate School of Pharmaceutical Sciences, The University of Tokyo
Introduction What is Aβ?
A senile plaque, one of characteristic brain pathological features of Alzheimer's disease (AD), is the brain parenchymal accumulation of abnormally aggregating insoluble proteins forming amyloid fibril abundant in β-sheet structure. Amyloid PET imaging, which has recently been developed to visualize senile plaques in the human brain, have shown that senile plaques appear approximately 10 to 15 years before the onset of cognitive dysfunction, indicating that senile plaques are the earliest lesion in AD. It was reported by Glenner et al. in 1984 and by Masters et al. in 1985 that the main component of senile plaques is amyloid β-peptide (Aβ), through the purification from cerebrovascular amyloid and senile plaques, respectively, and subsequent identification of amino acids.1,2)
Aβ is a peptide fragment consisting of approximately 40 to 43 amino acids. There are several Aβ species that vary in the N- or C-terminal, and the most common Aβ species in senile plaques are Aβ40, which consists of 40 amino acids and ends at the 40th Val residue, and Aβ42, which consists of 42 amino acids and ends at the 42nd Ala residue. Compared with Aβ40, Aβ42 is much more aggregative and accumulates earlier as senile plaque.3) In addition, some of deposited Aβ is modified, including Aβ with pyro-modification at the 3rd Glu residue.4)
Amyloid hypothesis
AD is mostly sporadic, but is only rarely autosomal dominant familial (familial AD, FAD). Genetic studies have identified the genes responsible for FAD to be APP, which encodes amyloid precursor protein (APP), and PSEN1 and PSEN2, which encode presenilin 1 and 2, catalytic subunit of the APP cleaving enzyme γ-secretase complex, respectively. These three genes have several FAD mutations that have been identified in families with FAD, and the mutations are reported to result in increased total Aβ production, increased relative production of aggregative Aβ42, and increased aggregation of Aβ itself. These results lead to "amyloid hypothesis," which assumes that aggregation and deposition of Aβ result in the development of AD.5) In addition, the A673T mutation in APP was identified as a protective mutation against AD, and this mutation was reported to result in decreased Aβ production,6) supporting the amyloid hypothesis as the mechanism underlying the development of AD.
The brain Aβ concentration is regulated through a balance between production and clearance, and an imbalance result in aggregation. This article describes Aβ from three aspects, that is, production, clearance, and aggregation, as well as the prospect for therapeutic strategies for AD targeting Aβ.
Process of Aβ production
Aβ is a peptide fragment released from its precursor protein APP. APP is a type I transmembrane protein and ubiquitously expressed in various tissues. Depending on the difference in splicing, APP has mainly three isoforms consisting of 695, 751, and 770 amino acids. One of them, APP695, which consists of 695 amino acids, is highly expressed in the brain.7,8,9)
As shown by the fact that Aβ is detected in the cerebrospinal fluid of healthy individuals, Aβ is not pathologically produced in the brain of AD patients, but is produced and secreted in the normal metabolic process of APP.10,11) Aβ is produced from APP through two-step cleavage (Fig. 1). First, the extracellular domain of APP is cleaved by β-secretase to secrete an extracellular fragment (sAPPβ). Subsequently, the remaining C-terminal fragment (C99) of APP is cleaved by γ-secretase to produce Aβ and AICD. Variations in the C-terminal of Aβ are introduced by γ-secretase cleavage; more specifically, several Aβ species from Aβ37 to Aβ49 are produced depending on the site of γ-secretase-cleaved amino acid.12) While highly produced Aβ40 and highly aggregative and toxic Aβ42 have traditionally attracted interest, Aβ43 is another highly aggregative and toxic Aβ species of recent interest.13) In addition, the development of γ-secretase modulators (GSMs), which modulate the cleavage activity of γ-secretase rather than inhibit cleavage, and studies about the mechanism of action of GSMs on the production of short Aβ species such as Aβ37 and Aβ38 are underway, and also attracts interest. BACE1 is the main β-secretase. On the other hand, γ-secretase is protein complex consisting of Aph-1, Pen-2, and nicastrin with presenilin 1 or 2 as catalytic subunit.
The metabolic process of APP includes the non-amyloidogenic pathway, in addition to the amyloidogenic pathway which produces Aβ (Fig. 1). The non-amyloidogenic pathway also involves two-step cleavage by α-secretase responsible for the first cleavage and by γ-secretase responsible for the second cleavage, like the amyloidogenic pathway. α-secretase cleaves the Aβ domain of APP between the 16th Lys residue and the 17th Leu residue to produce an approximately 3-kDa peptide fragment (p3) deficient of 16 amino acids in the N-terminal region of Aβ. ADAM10 is the main α-secretase in neurons.
It has recently been reported that Aβ production changes in a neural activity-dependent manner. In the mouse brain, it has been shown that brain area-specific differences in neural activity correlate with differences in Aβ production or accumulation, and that Aβ production decreases during sleep, when neural activity decreases.14,15) It has also been shown that long-term stimulation of neural activity using optogenetic technology results in increased Aβ accumulation.16) While there is no known mechanism underlying the neural activity-induced increase in Aβ production, increased neural activity may result in increased endocytosis, more specifically, increased endocytosis of APP on the cell surface, which, in turn, results in increased cleavage of APP by BACE1.
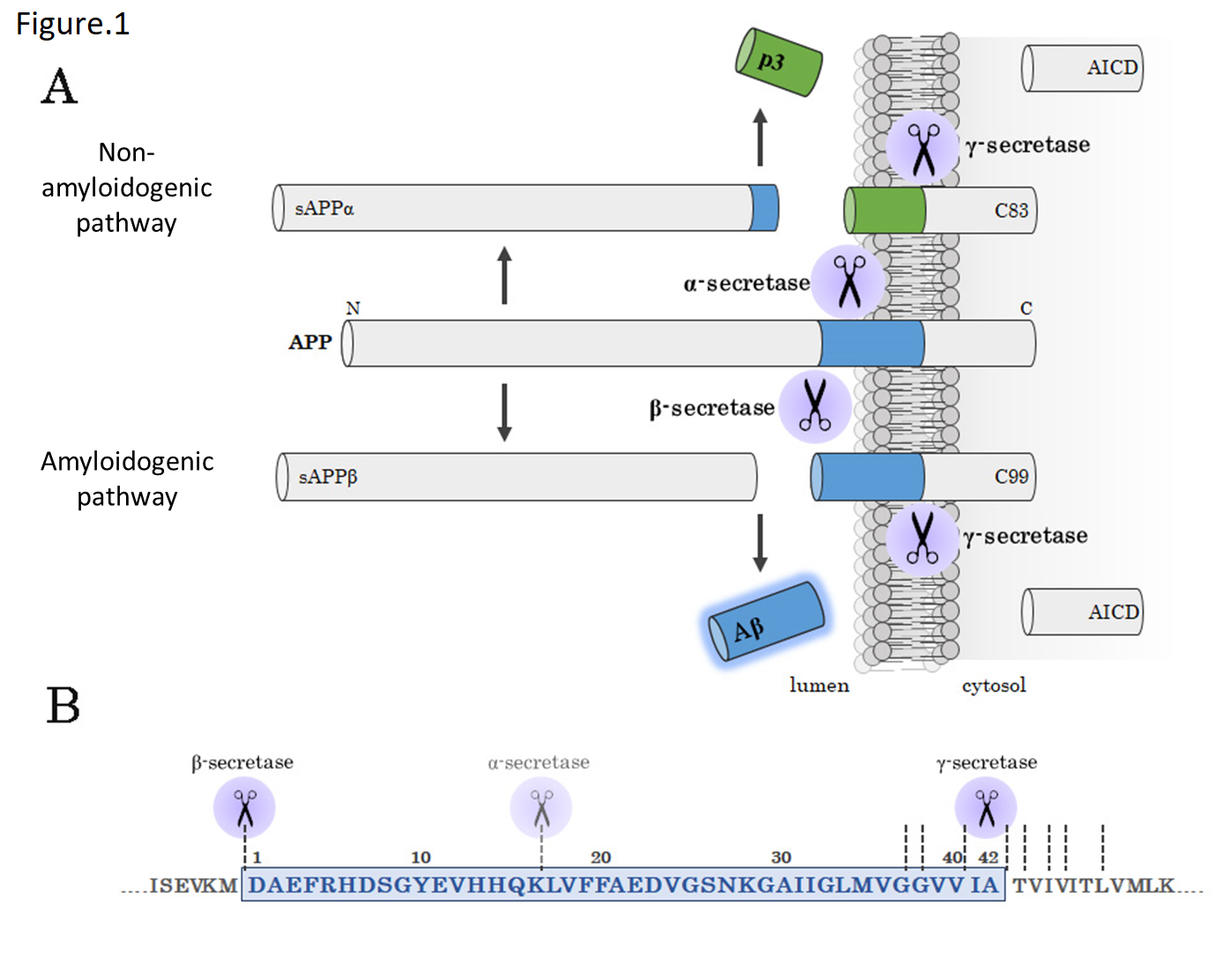
Fig 1. Amyloidogenic pathway and non-amyloidogenic pathway
(A) A schematic diagram of amyloidogenic pathway. APP is cleaved by β-secretase and γ-secretase to produce Aβ (amyloidogenic pathway) or cleaved by α-secretase and γ-secretase to produce p3 (non-amyloidogenic pathway).
(B) Aβ sequence and cleavage sites. The blue area represents the Aβ sequence.
Process of Aβ clearance
Usually, produced Aβ is rapidly cleared to maintain brain Aβ concentrations. However, some FAD patients develop AD due to disruption of this balance resulting from FAD mutation-induced increase in Aβ production. On the other hand, sporadic AD is shown to be associated with impaired Aβ clearance rather than increased Aβ production,17) suggesting the importance of studying the molecular mechanisms in detail of not only Aβ production, but also Aβ clearance. The main proposed mechanisms of Aβ clearance are degradation by Aβ-degrading enzymes, phagocytosis by glial and other cells, and efflux transport via the blood-brain barrier (BBB).
The representative Aβ-degrading enzymes are insulin-degrading enzyme (IDE) and neprilysin (NEP), which are metalloproteases, and kallikrein-related peptidase 7 (KLK7), which is a serine protease.18,19,20) Analyses in knockout mice have shown that all these enzymes degrade endogenous Aβ in the mouse brain, and decreased expression levels or degrading activities of the enzymes in the brain of AD patients have been reported. In addition, adeno-associated viral vector-mediated expression of NEP resulted in decreased Aβ accumulation,21) suggesting that these Aβ-degrading enzymes may serve as a therapeutic target for AD.
Aβ phagocytosis by glial cells such as microglia and astrocyte is another possible mechanism of Aβ clearance. Receptors that may be involved in Aβ phagocytosis, including the scavenger receptor, are expressed in microglia. Interestingly, moreover, several genes, which were found in a recent genome-wide association study (GWAS) to be possibly associated with AD,22) are highly expressed in microglia, suggesting that glial cells may be involved in the development of AD in some way. In fact, CD33 and TREM2, among these genes, have been reported to affect Aβ clearance.23,24).
Process of Aβ aggregation
Aβ has no characteristic structure when existing as a monomer, but forms amyloid fibrils abundant in β-sheet structure when aggregating. Aβ42 is more aggregative than Aβ40 and the earliest Aβ species that accumulates in the brain as senile plaque. While it is not clear why Aβ42 is more susceptible to structural changes than Aβ40, some say that structural stabilization of the C-terminal due to the β-hairpin structure at the 31st to 34th residues and the 38th to 41st residues may facilitate structural change into a β-sheet structure.27) In addition, amyloid fibrils may form a β-sheet structure with the turn structure at the 25th to 29th residues of the Aβ molecule and form a cross β-sheet structure through intermolecular interactions.25,26) Since the region forming an intramolecular turn structure is a hot spot of FAD genetic mutations resulting in increased Aβ aggregation, these mutations may promote structural changes of Aβ, supporting the possible acceleration of fibril formation from the structural aspect as well.
In in vitro experiments, a two-process model consisting of nucleation and elongation is proposed for Aβ amyloid formation.28,29) The nucleation process is the first process in which Aβ monomers form seeds that serve as a nucleus for fibril growth while undergoing structural changes; this process is slow and a rate-limiting step for amyloid formation. The nucleation process occurs earlier for Aβ42 than for Aβ40. The subsequent elongation process is a rapid process in which Aβ molecules bind to the seeds while undergoing structural changes, resulting in fibril growth. In vitro, Aβ40 aggregation is slow when it is incubated alone, but markedly accelerated when it is incubated with a small quantity of fibrotic Aβ. Also in vivo, injection of brain lysate derived from AD patients or AD model mice with Aβ deposition into the brain of young mice with no Aβ deposition results in accelerated Aβ accumulation due to injected Aβ acting as seed,30) suggesting the importance of controlling seed formation. Interestingly, a structural analysis of Aβ amyloid derived from the brain of AD patients showed that the amyloid structure varied each patient, suggesting that the amyloid structure may differ depending on the disease progression, and thus suggesting the importance of the first seed structure.26)
The original amyloid hypothesis assumed that Aβ accumulation as senile plaque leads to the development of AD. However, without a clear correlation between senile plaque and reduced cognitive function, as well as given the finding that Aβ oligomers, which are intermediates in aggregation, are more toxic than amyloid fibrils, the amyloid hypothesis is partially corrected with the statement that Aβ oligomers are particularly toxic. Aβ oligomers are defined as soluble low-molecular-weight Aβ polymers, unlike insoluble Aβ forming an amyloid fibrous structure. In vitro experiments have reported various Aβ oligomers, including paranuclei, protofibrils, and Aβ-derived diffusible ligands (ADDLs).31,32,33) In vivo, low-n oligomers such as dimer and trimer, as well as 56-kDa 12-mer Aβ*, have been identified to date.34,35) Since these Aβ oligomers are reported to have high synaptic toxicity and cytotoxicity, the mechanism of formation and nature of them are interesting.
Therapeutic strategies for AD targeting Aβ
Based on the amyloid hypothesis, various therapeutic strategies for AD targeting Aβ have been proposed. While studies have been underway primarily to inhibit Aβ production, promote Aβ clearance, and inhibit Aβ aggregation, and several candidate compounds have been developed at the basic experimental level, the following two points of view being tested in clinical studies are described here:
One is to control secretase activities with an aim to inhibit Aβ production. However, γ-secretase cleaves various substrates in addition to APP, and side effects due to inhibition of Notch cleavage are great problems. It is therefore desirable to develop GSMs, which modulate the cleavage activity of γ-secretase to reduce Aβ42 production specifically, rather than γ-secretase inhibitors. On the other hand, since β-secretase BACE1 knockout mice are not phenotypically as abnormal as presenilin knockout mice, and β-secretase-induced cleavage is a rate-limiting step in the process of Aβ production, BACE1 inhibitors may be promising drugs for AD.
The other is to promote Aβ clearance. In this regard, anti-Aβ antibodies as passive immunotherapy, and Aβ vaccines as active immunotherapy, have been tested in clinical studies. In 1999, Schenk et al. reported that Aβ accumulation in the brain decreased in APP transgenic mice immunized with Aβ aggregates.36) Later, it was reported that a similar effect was observed after administration of anti-Aβ antibodies,37) showing that anti-Aβ antibodies promote Aβ clearance. While a precise mechanism of Aβ clearance has yet to be determined, activated Aβ phagocytosis by microglia and increased peripheral sink of Aβ (sink hypothesis) have been proposed. It should be noted that, given minimal distribution of antibodies to the brain and epitope-specific differences in the efficacy of antibodies, a detailed mechanism has yet to be elucidated.
However, the results of clinical studies of these drugs targeting Aβ have been disappointing. One reason may be delayed start of treatment. Since senile plaques begin to accumulate 10 to 15 years before the onset of cognitive dysfunction as mentioned above, a therapeutic strategy targeting only Aβ may not always improve symptoms of pathologically advanced AD with cognitive dysfunction. Accordingly, preemptive treatment, that is, preventive intervention in the asymptomatic stage with Aβ accumulation or the preclinical stage corresponding to early AD, is being developed.38,39) To make this therapeutic strategy effective, methods for early diagnosis of Aβ accumulation, including simple diagnostic modalities and biomarkers of Aβ amyloid, must also be developed.
Conclusion
Many years of basic studies in AD generally validate the amyloid hypothesis attributing the development of AD to Aβ as well as the therapeutic strategy targeting Aβ. However, since no curative treatment for AD has been established, it is desired to elucidate and clinically apply a detailed molecular mechanism of the contribution of Aβ to AD.
simple diagnostic modalities and biomarkers of Aβ amyloid, must also be developed.
References
- Glenner, G. G. et al . : Biochem. Biophys. Res.Commun ., 120, 885-890(1984)
- Masters, C. L. et al . : Proc. Natl. Acad. Sci. USA, 82, 4245-4249(1985)
- Iwatsubo, T. et al . : Neuron , 13, 45-53(1994)
- Saido, T. C. et al . : Neuron , 14, 457-466(1995
- Selkoe, D. J. and Hardy, J. : EMBO Mol. Med ., 8, 595-608(2016)
- Jonsson, T. et al . : Nature , 488, 96-99(2012)
- Ponte, P. et al . : Nature , 331, 525-527(1988)
- Tanzi, R. E. et al . : Nature , 331, 528-530(1988)
- Kitaguchi, N. et al . : Nature , 331, 530-532(1988)
- Haass, C. et al . : Nature , 359, 322-325(1992)
- Seubert, P. et al . : Nature , 359, 325-327(1992)
- Qi-Takahara, Y. et al . : J. Neurosci ., 25, 436-445(2005)
- Saito, T. et al . : Nat. Neurosci ., 14, 1023-1032(2011)
- Kang, J. E. et al . : Science , 326, 1005-1007(2009)
- Bero, A. W. et al . : Nat. Neurosci ., 14, 750-756(2011)
- Yamamoto, K. et al . : Cell Rep ., 11, 859-865(2015)
- awuenyega, K. G. et al . : Science , 330, 1774(2010)
- Iwata, N. et al . : Nat. Med ., 6, 143-150(2000)
- Qiu, W. Q. et al . : J. Biol. Chem ., 273, 32730-32738(1998)
- Kidana, K. et al . : unpublished
- Iwata, N. et al . : Sci. Rep ., 3, 1472(2013)
- Lambert, J. C. et al . : Nat. Genet ., 45, 1452-1458(2013)
- Griciuc, A. et al . : Neuron , 78, 631-643(2013)
- Wang, Y. et al . : Cell , 160, 1061-1071(2015)
- Petkova, A. T. et al . : Proc. Natl. Acad. Sci. USA, 99, 16742-16747(2002)
- Lu, J. X. et al . : Cell , 154, 1257-1268(2013)
- Sgourakis, N. G. et al . : J. Mol. Biol ., 368, 1448-1457(2007)
- Come, J. H. et al . : Proc. Natl. Acad. Sci. USA, 90, 5959-5963(1993)
- Jarrett, J. T. et al . : Biochemistry , 32, 4693-4697(1993)
- Meyer-Luehmann, M. et al . : Science , 313, 1781-1784(2006)
- Walsh, D. M. et al . : J. Biol. Chem ., 272, 22364-22372(1997)
- Bitan, G. et al . : Proc. Natl. Acad. Sci. USA , 100, 330-335(2003)
- Lambert, M. P. et al . : Proc. Natl. Acad. Sci. USA, 95, 6448-6453(1998)
- Shankar, G. M. et al . : Nat. Med ., 14, 837-842(2008)
- Lesne, S. et al . : Nature , 440, 352-357(2006)
- Schenk, D. et al . : Nature , 400, 173-177(1999)
- Bard, F. et al . : Nat. Med ., 6, 916-919(2000)
- Sperling, R. et al . : Neuron , 84, 608-622(2014)
- Reiman, E. M. et al . : Nat. Rev. Neurol ., 12, 56-61(2016)