Vol. 4. Molecular Imaging in Alzheimer's Disease
This article was written by Dr. Makoto Higuchi, National Institute of Radiological Sciences, National Institutes for Quantum and Radiological Science and Technology.
Introduction
Alzheimer's disease (AD) is neuropathologically characterized by the deposition of protein aggregates and progressive neuronal death in the brain. Pathological protein fibrils deposited in the brain of patients with AD are primarily composed of amyloid β (Aβ) and tau. Researchers have proposed an etiological cascade initiated by Aβ and tau accumulations in the brain, which triggers sequential pathological events such as activation of inflammatory glial cells and abnormal neurotransmissions, resulting in the neuronal death (Fig. 1).1) Visualization of these events in the living brain could help to clarify the spatiotemporal profiles of pathogenetic processes characteristic of AD, offering indices for the diagnosis and staging of the disease. Until the 21st century, pivotal imaging techniques assisting the clinical diagnosis of AD were CT and MRI, which were used for the assessments of brain atrophy and cerebrovascular lesions, single-photon emission computed tomography (SPECT), which was used for examining regional cerebral blood flow, and positron emission tomography (PET) with fluorodeoxyglucose as a tracer, which was used for assays of the glucose metabolism in the brain. While these techniques still provide information facilitating the diagnosis by detecting aberrant neurotransmissions (and resultant alterations of brain activities) and neuronal death located relatively downstream in the aforementioned cascade, they are unable to capture pathological processes at a very early stage. Since Aβ and tau depositions in the brain, which are core lesions of AD, may precede the clinical onset by approximately 20 years, imaging of these changes enables a neuropathology-based diagnosis in an asymptomatic phase. In addition, initiation of an intervention in the Aβ and tau pathologies at an optimal time point based on the imaging of these depositions is expected to decelerate the pathological progression, leading to the prevention and therapy of the disease.
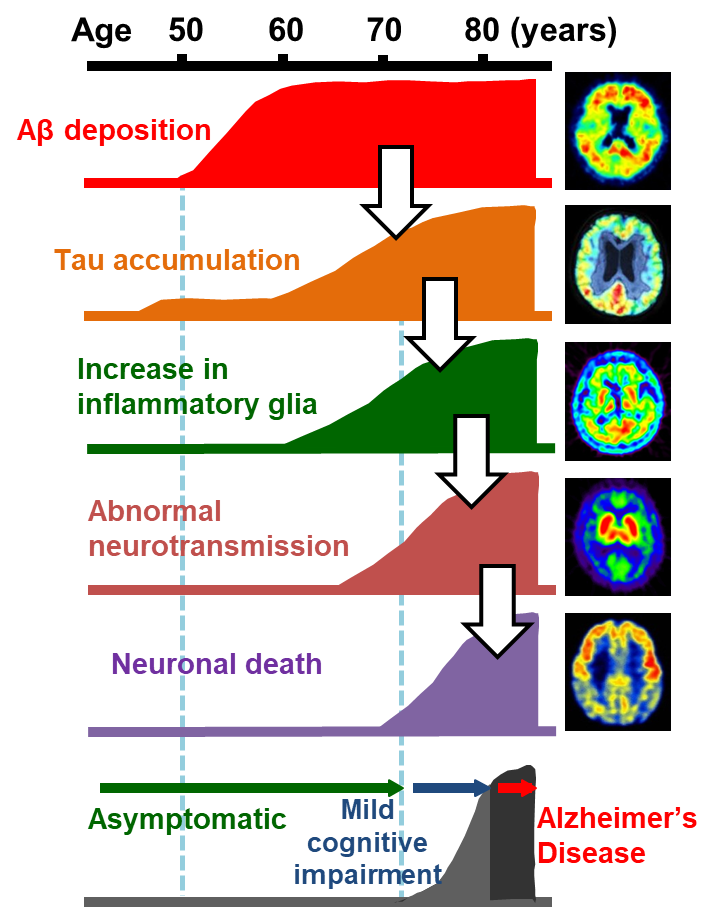
PET and MRI technologies allow visualization of pathogenetic components of the cascade in the living brain.
Imaging of Aβ lesions
Senile plaques, extracellular deposits of Aβ, result from the fibrillization of Aβ molecules mediated by the formation of a β-sheet secondary structure. Thioflavin T, a fluorescent dye, is known as a traditional small-molecule compound that binds to the β-pleated sheet. Since thioflavin T hardly penetrates the blood-brain barrier (BBB) due to its high polarity attributable to a quaternary amine within it, a neutral derivative of thioflavin T was developed by University of Pittsburgh to increase the efficiency of the trans-BBB entry to the brain and was dubbed Pittsburgh Compound B (PiB).2) PiB labeled with 11C, a positron-emitting radioisotope, was used as a tracer in clinical PET studies and was reported by a work published in 2004 to be useful for imaging Aβ depositions in the brains of patients with AD.2) While 11C-labeled PiB is not widely used due to its short radioactivity half-life approximating 20 minutes, Aβ tracers labeled with 18F, which has a half-life of approximately 110 minutes, have been developed, and several radiofluorinated compounds are currently available for clinical examinations as deliverable PET imaging agents.
PET studies of Aβ pathologies have hitherto provided the following major findings3,4): (1) PET detects Aβ deposits in a subset of cognitively normal elderly individuals, and the frequency of possessing PET-visible Aβ plaques increases with age; (2) normal elderly individuals and patients with mild cognitive impairment (MCI) who have PET-detectable Aβ accumulation are more likely to proceed to MCI and AD, respectively, than those without noticeable Aβ depositions; (3) since the binding of a PET probe to Aβ plaques plateaus at an MCI stage, Aβ PET may not offer an indicator of further disease progressions (Fig. 2); (4) the spatial mismatch between increased Aβ PET signals and brain atrophy indicates that Aβ accumulation per se does not provoke significant neuronal loss; and (5) PET detects Aβ deposition in more than half of patients with Lewy body dementia, in addition to patients with AD.
According to these observations, the significances of Aβ PET in healthy elderly individuals, patients with MCI and dementia are summarized in Table 1. Negative imaging findings are associated with conclusive implications, whereas positive imaging findings direct to more speculative indications. In particular, future longitudinal studies will determine whether Aβ depositions in the elderly with no cognitive impairment may correspond to very early AD. The coverage of Aβ PET by a health insurance system is not yet established in many countries and is primarily applied to exclusive diagnosis based on negative imaging findings in a small number of nations. This restriction is chiefly attributable to the lack of therapies counteracting Aβ depositions, along with insufficient evidence for the utility of Aβ PET in the therapeutic decision-making. Until now, no clinical trials of anti-Aβ therapies, including Aβ vaccines, have led to approval by a regulatory agency, and the need to start treatment for individuals with no or minimal cognitive declines has been supported.5) PET imaging of Aβ plaques would be suitable for identifying these subjects at a preclinical AD stage to be recruited for clinical trials.
PET imaging of Aβ depositions may serve not only clinical but also basic research and development. Indeed, transgenic mice genetically modified with a gene encoding the amyloid precursor protein, which induces Aβ aggregations in the brain, Aβ PET was employed for elucidating differences in the properties of Aβ lesions between humans and the model mice, as along with analyses of the relationships among Aβ pathologies, neuroinflammation, and neuronal dysfunctions (Fig. 3).6) In model mice, imaging of individual Aβ lesions has been enabled by intravital two-photon laser fluorescence microscopy following systemic administration of highly fluorescent PiB derivatives and other related ligands (Fig. 3).7) The in-vivo optical and PET imaging also helps to quantify the rate of the Aβ fibrillization in the living brain, which could be utilized for non-clinically assessing efficacies of anti-Aβ therapies exemplified by vaccines.6,7)
| Positive | Negative | |
|---|---|---|
| Dementia | Probable AD or Lewy body dementia (other forms of dementia are unlikely) |
Non-AD dementia (other forms of dementia) |
| Mild cognitive impairment | Likely to progress to AD within a few years |
Almost unlikely to progress to AD within a few years |
| No cognitive impairment | Likely to progress to AD within 5 to 10 years (verification is required) |
Almost unlikely to progress to AD within 5 to 10 years |
Table 1. Significance of positive and negative findings in Aβ PET examination
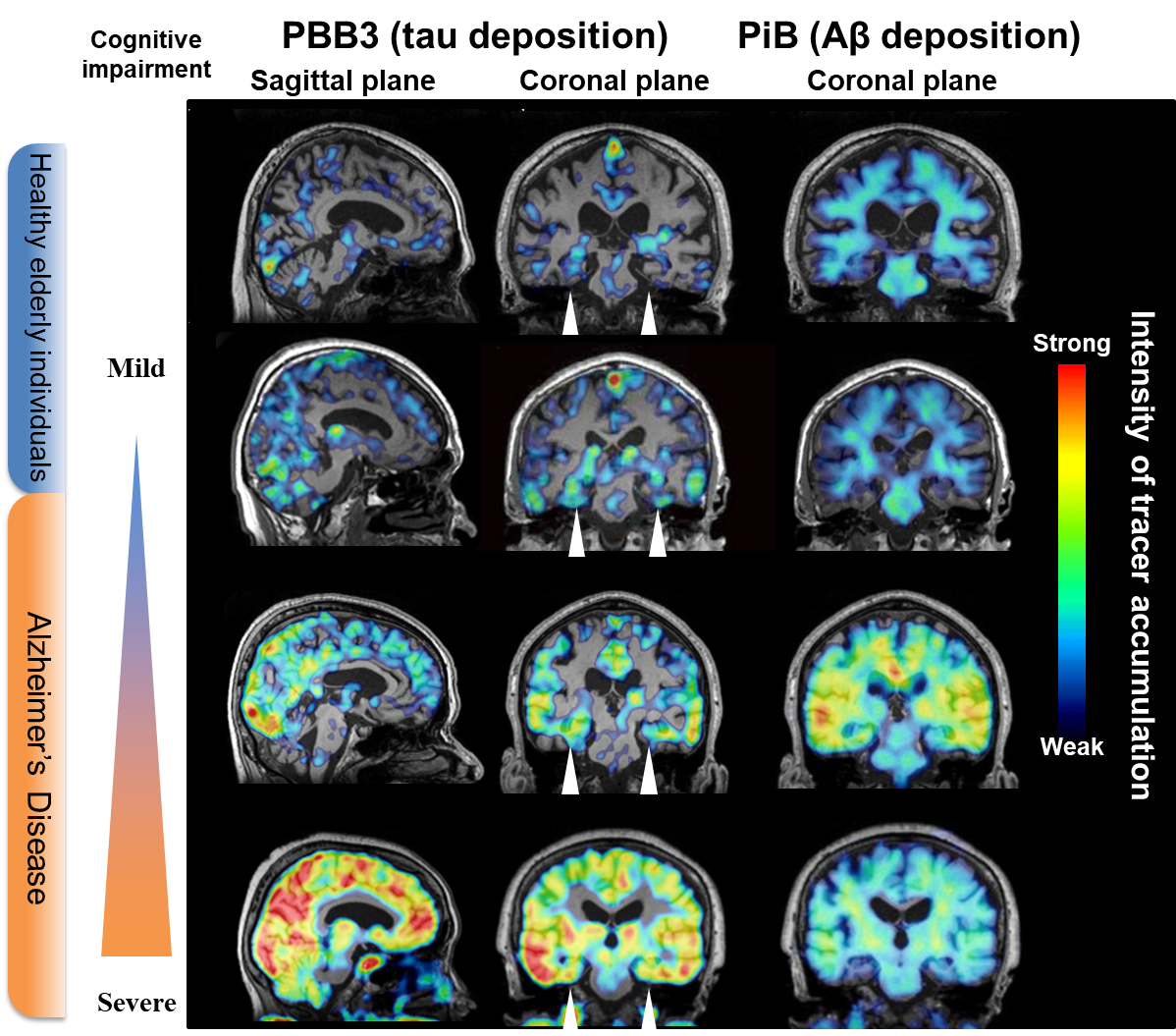
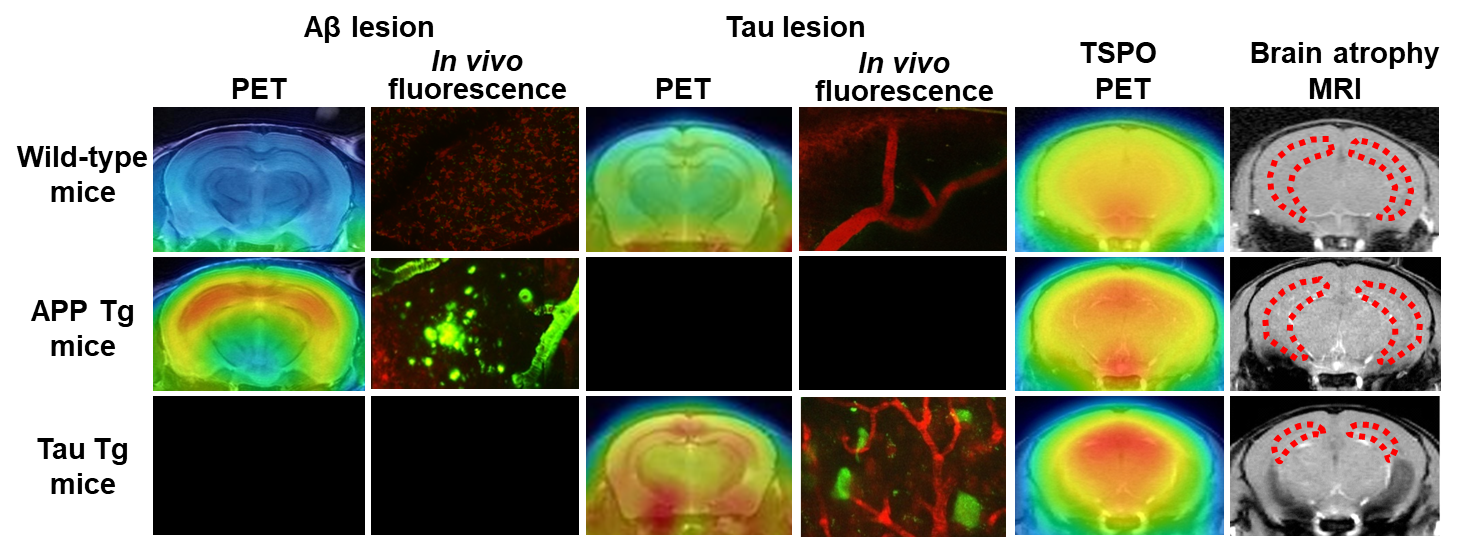 Figure 3. Bioimaging of Aβ and tau depositions, neuroinflammation, and brain atrophy in model mice
Figure 3. Bioimaging of Aβ and tau depositions, neuroinflammation, and brain atrophy in model miceIn tau transgenic (Tg) mice, tau deposits are noted along with increased levels of TSPO, a marker of inflammatory gliosis. MRI shows atrophy of the hippocampus (dotted area) and several other areas. By contrast, Aβ pathologies are noticeable without overt hippocampal atrophy and with little evidence of TSPO-positive aggressive microgliosis.
Imaging of tau lesions
The failure of clinical trials for anti-Aβ therapies gave rise to a view that the deposition of tau rather than Aβ could be a target of efficacious disease-modifying treatments. Based on this concept, pharmaceutical companies and research institutes are also developing anti-tau therapies, including tau vaccines and tau aggregation inhibitors.5) Tau inclusions are known to form in the AD brains accompanying Aβ pathologies and in the brains of non-AD neurodegenerative dementias such as frontotemporal lobar degeneration lacking Aβ depositions.8) A notable feature of tau lesions is their close association with neurodegeneration in these disorders collectively referred to as tauopathies.8) Since familial neurodegenerative diseases caused by mutations in the tau gene also provoke tau accumulations and neuronal loss without Aβ deposits, tau pathologies may be mechanistically linked to neurodegeneration driving neurons to toxic death.8) In transgenic mice genetically modified with a human tau gene having the aforementioned mutations, significant neuronal death in addition to tau aggregations is observed, supporting the relations of tau abnormalities to the neurotoxicity.9) By contrast, there is no profound neuronal loss in the amyloid precursor transgenic mice, suggesting that pathological Aβ assemblies are less neurotoxic than intraneuronal tau multimers (Fig. 3).
It is accordingly necessary to visualize tau lesions in the living brain for the development of anti-tau therapies as well as the clarification of roles played by tau abnormalities in the neurodegenerative pathogenesis, since this technology allows researchers and clinicians to identify the optimal time point of therapeutic intervention based on the quantity and extent of tau deposits, and to evaluate the treatment outcomes in an objective fashion. In response to these demands, PET imaging agents have been developed to detect tau lesions in the brains of living subjects. While tau, like Aβ, self-aggregates in a β-sheet structure, compounds that selectively bind to the β-sheet of tau versus Aβ aggregates have been generated and applied to clinical PET since the early 2010s (Table 2). Representative tau PET tracers include AV-1451 (also known as T807 or flortaucipir) developed by Avid Radiopharmaceuticals,10) THK-5351 developed by Tohoku University and clinically evaluated by General Electric,11) and PBB3 developed by our group at National Institute of Radiological Sciences.12) Tau PET studies with these radioprobes have then provided the following findings (Fig. 2): (1) Tau fibrils are deposited in the hippocampal formation of a subpopulation of elderly individuals with normal cognitive functions, in a manner independent of Aβ depositions; (2) Aβ pathologies may accelerate the spread of tau depositions from the hippocampal formation to the entire limbic system and subsequently to the neocortex. The spatial extent of tau accumulations is intimately correlated with the progression of MCI and AD and may therefore serve as an objective index of the disease severity; and (3) The high regional concordance between tau lesions and brain atrophy in MCI and AD suggests on-site neurotoxicities of fibrillar tau species.
In addition to imaging of the tau fibrillogenesis in normal aging and AD-spectrum disorders as described above, it is of critical importance to capture tau lesions in non-AD dementias for diagnostic and therapeutic purposes. It should be noted that tau proteins are constituted of six isoforms in the brain, and all isoforms are incorporated in the AD-type deposits. By contrast, major forms of frontotemporal lobar degeneration syndromes are characterized by depositions of only '3-repeat' isoforms with three repeat domains as represented by Pick's disease, or "4-repeat" isoforms with four repeat domains as exemplified by progressive supranuclear palsy and corticobasal degeneration.8) These differences in isoform composition may result in ultrastructural diversities of tau assemblies, giving rise to distinct reactivities of tracer compounds. Among the currently available tau PET probes, AV-1451 is shown to be more selective for AD-type tau aggregates, while it binds weakly to tau lesions consisting of 3-repeat or 4-repeat isoforms only.13) PBB3 is documented to possess a high affinity for tau lesions with all isoform compositions, but the contrast for tau pathologies yielded by PBB3 is not as high as AV-1451 and allied tracers because of its greater susceptibility to biometabolism.12,13) In addition, labeling with 11C, rather than 18F, limits the utility of PBB3 as a diagnostic agent in a clinical setting. Since THK-5351 binds not only to tau fibrils but also to monoamine oxidase B, and this enzyme is abundantly expressed in reactive astrocytes, it is not clear whether increased radiosignals reflects tau deposition or activation of astrocytes, resulting in the interruption of clinical development of this radioprobe.
Next-generation tau PET tracers are currently being developed, and these compounds yield high contrasts for tau deposits without binding to non-tau molecules such as monoamine oxidases in the brain parenchyma, and are labeled with 18F for the wide availability. Moreover, PI-2620 developed by Piramal Imaging and PM-PBB3 developed by National Institute of Radiological Sciences14) have been shown to bind to both AD and non-AD tau lesions (Table 2). PET imaging with these radioprobes will accordingly offer powerful tools in selecting subjects with diverse tauopathies for clinical studies of anti-tau therapies and assessing the capability of such treatments for the suppression of the pathological progression.
Among the known tau tracers, PBB3 and PM-PBB3, which can react with tau lesions in tauopathy model mice as well, are applicable to a longitudinal PET analysis of tau pathologies over time and multimodal PET and MRI assessments of the relationship between tau depositions and neuroinflammation or brain atrophy (Fig. 3).12,14,15) As these chemicals are fluorescent molecules, they can be utilized for monitoring the formation of individual tau with a two-photon laser fluorescence microscope in vivo (Fig. 3).12,14) These multiscale imaging assays in model animals reinforce the non-clinical development of anti-tau therapeutics.
| PET Tracer | Developer | Contrast for AD tau detection |
Binding to non-Alzheimer tau lesion |
Off-target binding | |
|---|---|---|---|---|---|
| MAO | Choroid plexus | ||||
| [11C] PBB3 | National Institute of Radiological Sciences |
1.2 - 1.4 | 4-repeat tau lesion 3-repeat tau lesion |
- | ± |
| [18F] AV-1451 | Avid/Lilly | 1.3 - 1.6 | 4-repeat tau lesion (low contrast) |
MAO-A | + |
| [18F] THK-5351 | Tohoku University | 1.4 - 1.8 | 4-repeat tau lesion | MAO-B | - |
| [18F] PI-2620 | Piramal | 2.0 - 2.5 | 4-repeat tau lesion | - | - |
| [18F] GTP1 | Genentech | 2.0 - 3.0 | - | - | |
| [18F] RO6958948 | Roche | 2.0 - 2.5 | - | - | |
| [18F] MK-6240 | Merck | 2.0 - 3.0 | - | - | |
| [18F] PM-PBB3 | National Institute of Radiological Sciences |
2.0 - 2.5 | 4-repeat tau lesion 3-repeat tau lesion |
- | + |
Table 2. Clinically available tau PET tracers
Sources are data from articles and conference presentations. "Next-generation" tracers are shown in red. The contrast for tau detection is expressed as the ratio of the cerebral cortical tracer uptake between patients with early (or preclinical) AD and normal controls.
MAO is an abbreviation for monoamine oxidase.
Imaging of other pathology and future prospects
As described above, Aβ and tau depositions, the most upstream processes in the pathogenetic cascade displayed in Figure 1, have been successfully visualized by PET and intravital fluorescence imaging. While inflammatory gliosis is considered to be a key intermediate event linking deposition of the fibrillary protein aggregates and neuronal deteriorations, it is yet to be determined when reactive glial cells emerge in the course of the disease progression. Moreover, whether activated glial cells deleteriously or protectively act on neurons still remains elusive. In light of the fact that microglia with an aggressive phenotype exhibit elevated levels of mitochondrial translocator protein (TSPO),16) PET imaging of inflammatory microgliosis in the brain has been enabled by a small-molecule tracer for TSPO.16,17) The role of reactive astrocytes and microglia in the disease can be clarified by applying PET to the investigation of associations among TSPO-positive gliosis, accumulations of Aβ and tau, and brain atrophy in animal models and humans. Furthermore, changes in glial phenotypes as assessed by TSPO-PET would be utilized as indicators of therapeutic efficacies in the development of immunomodulating drugs such as vaccines.
In addition to clinical PET imaging, PET imaging of TSPO in a mouse model of dementia has been realized (Fig. 3),16,18) identifying distinct glial phenotypes provoked by Aβ and tau depositions. Both TSPO-positive aggressive microglia and brain atrophy due to neuronal death are observed in tau transgenic mice, but not in amyloid precursor transgenic mice (Fig. 3), suggesting that TSPO-positive microglia are tightly linked to the neurodegenerative tau accumulations leading to neuronal death.16,18) Mutual relationships among Aβ and tau depositions, activation of TSPO-positive microglia, and neuronal death (brain atrophy) can be compared between humans and model mice using imaging techniques commonly shared by these species to evaluate the translatability of findings in model mice to humans and to assess the validity of animal models of the disease based on the similarities of their etiological pathways to those of humans.
References
- Higuchi, M. et al. : Biochim Biophys. Acta, 1802, 373 (2010).
- Klunk, W. E. et al. : Ann. Neurol., 55, 306 (2004).
- Villemagne, V. L. et al. : Lancet Neurol., 12, 357 (2013).
- Weiner, M. W. et al. : Alzheimers Dement., 11, e1-120, (2015).
- Giacobini, E. and Gold, G. : Nat. Rev. Neurol., 9, 677 (2013).
- Maeda, J. et al. : J. Neurosci., 27, 10957 (2007).
- Bacskai, B. J. et al. : Nat. Med., 7, 369 (2001).
- Higuchi, M. et al. : Neuromolecular Med., 2, 131 (2002).
- Yoshiyama, Y. et al. : Neuron, 53, 337 (2007).
- Johnson, K. A. et al. : Ann. Neurol., 79, 110 (2016).
- Harada, R. et al. : J. Nucl. Med., 57, 208 (2016).
- Maruyama, M. et al. : Neuron, 79, 1094 (2013).
- Ono, M. et al. : Brain, 140, 764 (2017).
- Tagai, K. et al. : medRxiv, DOI: 10.1101/2020.03.05.20028407 (2020).
- Ishikawa, A. et al. : J. Alzheimer's Dis., 61, 1037 (2018).
- Ji, B. et al. : J. Neurosci., 28, 12255 (2008).
- Kreisl, W. C. et al. : Brain, 136, 2228 (2013).
- Maeda, J. et al. : J. Neurosci., 31, 4720 (2011).