4th review: Macrophages control the inflammation and subsequent neural repair after ischemic stroke
This article was written by Dr. Kento Otani1,2, and Dr. Takashi Shichita1,3.
1 Stroke Renaissance Project, Tokyo Metropolitan Institute of Medical Science, and Core Research for Evolutional Science and Technology (CREST), Japan Agency for Medical Research and Development (AMED),
2 Division of Biochemistry, Faculty of Pharmacy and Graduate School of Pharmaceutical Science, Keio University
3 Precursory Research for Innovative Medical Care (PRIME), Japan Agency for Medical Research and Development (AMED)

Introduction
Cerebrovascular disease (stroke) is a major cause of chronic functional deficits, such as being bedridden. Ischemic stroke comprises approximately 80% of cerebrovascular disease in Japan. In ischemic stroke, decreased cerebral blood flow causes ischemic necrosis of brain tissue due to the deprivation of nutrients and oxygen, leading to severe cerebral inflammation. Cerebral ischemia induces the breakdown of the blood-brain barrier (BBB) and the infiltration of immune cells such as macrophages and neutrophils into the brain tissue. Macrophages are major producers of inflammatory cytokines and play major roles in the inflammation immediately after the onset of ischemic stroke. The function of infiltrating macrophages changes from an inflammatory phenotype to a reparative phenotype within several days of the onset of ischemic stroke. As two previous review articles have described the important role of microglia, in this review we describe the function of macrophages, another important myeloid cell in the brain, and the cellular and molecular mechanisms underlying cerebral post-ischemic inflammation and neural repair by macrophages. We also discuss the possibility of therapeutic drug development targeting macrophage function in ischemic stroke.
The role of macrophages in the inflammation after ischemic stroke
Circulating macrophages infiltrate the ischemic brain when the BBB is broken down. C-C chemokine receptor 2 (CCR2) is an important chemokine receptor for the infiltration of macrophages into the ischemic brain1). C-C motif chemokine 2 (CCL2), also called monocyte chemoattractant protein 1 (MCP1), is produced by ischemic brain cells and functions as a ligand for CCR2. CCR2 deficiency in monocytes attenuates the infiltration of circulating monocytes and macrophages into ischemic brain tissue2). Thus, the CCL2-CCR2 signal is considered to be essential for macrophages to infiltrate into the brain.
Ischemic cell death induces the release of endogenous self molecules within brain cells into the extracellular space, which are called damage-associated molecular patterns (DAMPs)3). DAMPs that are recognized by pattern recognition receptors (PRRs) in immune cells trigger cerebral post-ischemic inflammation by inducing the production of inflammatory cytokines and chemokines (Fig. 1). To date, many kinds of DAMPs have been identified, such as nucleic acids, proteins, and phospholipids. Among these, high mobility group box1 (HMGB1) and peroxiredoxin (PRX) family proteins are major DAMPs in the ischemic brain.
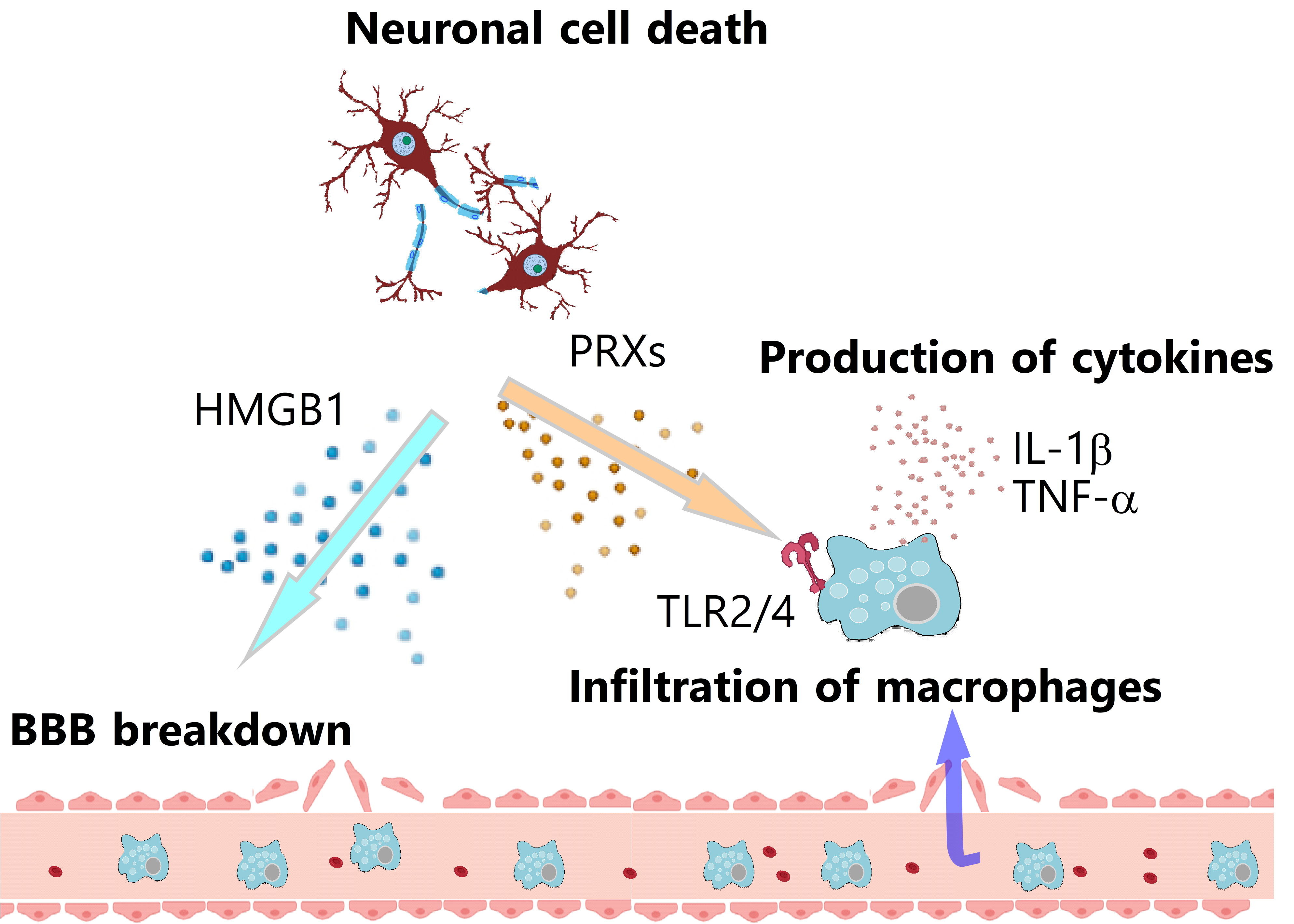 Figure 1: Induction of inflammation after ischemic stroke
Figure 1: Induction of inflammation after ischemic stroke
HMGB1, the DNA-binding protein that exists in the nucleus of brain cells, is released extracellularly from ischemic necrotic brain cells 2 to 4 h after the onset of ischemic stroke7). In ischemic brain tissue, HMGB1 induces the breakdown of the BBB, which leads to the drastic infiltration of various immune cells8). HMGB1 increases the expression of matrix metalloproteinase 9 (MMP9) in ischemic brain tissue, which promotes BBB disruption and cerebral inflammation9). HMGB1 also activates neutrophils to induce the release of extracellular traps, which are a complex of genomic DNA and bacteriocidal enzymes10). The formation of these neutrophil extracellular traps (NETs) is observed not only in infarcted brain tissue but also within cerebral blood vessels. The NETs in ischemic brain tissue are considered to promote cerebral post-ischemic inflammation and ischemic neuronal injuries. It is of note that the occlusion or severe stenosis of the cerebral artery stagnates cerebral blood flow and induces the release of intravascular NETs, which may promote the formation of thrombi and accelerate the ischemic stroke pathologies by further decreasing the cerebral blood flow around the brain infarct.
PRXs were originally known as the antioxidant proteins within brain cells that metabolize hydrogen peroxide into water and protect ischemic brain cells from oxidative stress11). The function of intracellular PRXs is thus neuroprotective, and these intracellular PRXs are released into the extracellular space if ischemic stresses result in necrotic brain cell death. The extracellularly released PRXs activate the infiltrating macrophages to induce the expression of inflammatory cytokines. Toll-like receptors (TLRs) are important for activating the immune cells, given that deficiency in Myd88, the essential adaptor protein of the TLR signaling pathway, markedly decreases the inflammatory cytokine expression in the infiltrating myeloid cells12). TLRs are one of the PRRs that recognize the molecules derived from pathogens to trigger immune responses; however, TLRs also recognize self molecules released from damaged tissues. In fact, the extracellularly released PRXs directly activate TLR2 and TLR4 to induce the expression of inflammatory cytokines, such as interleukin-1b (IL-1β), IL-23, and tumor necrosis factor (TNF)-α in infiltrating macrophages. These inflammatory cytokines promote cerebral post-ischemic inflammation that exacerbates brain edema and induces secondary ischemic neuronal damage, leading to worsened neurological deficits and poor functional prognosis in rodent models of ischemic stroke and in human stroke patients13).
The important roles of macrophages in the resolution of cerebral post-ischemic inflammation
Macrophage infiltration and the subsequent inflammation are closely related to ischemic stroke pathologies and functional prognosis. Macrophage infiltration also contributes to the resolution of inflammation and neural repair after ischemic stroke2). The expression of inflammatory cytokines reaches its peak around several days after stroke onset and decreases thereafter14). Infiltrating macrophages and reactive glial cells also produce IL-10 and transforming growth factor b (TGF-β), the major anti-inflammatory molecules, in the ischemic brain. Overexpression of IL-10 by adenoviral transduction in the ischemic brain shows neuroprotective effects against global brain ischemia15). TGF-β is an important factor in the induction of anti-inflammatory immune cells, such as regulatory T cells, and the repair of ischemic brain tissue16). The endogenous cellular and molecular mechanisms underlying the resolution of inflammation and subsequent neural repair are considered to exist in the ischemic brain.
The removal of DAMPs in the ischemic brain is necessary for the resolution of cerebral post-ischemic inflammation. Class A scavenger receptors, MSR1 and MARCO, in the infiltrating macrophages are important for recognizing and internalizing DAMPs, such as PRXs and HMGB1. Deficiency in MSR1 and MARCO in the infiltrating macrophages delays the clearance of DAMPs from ischemic brain tissue, resulting in prolonged inflammation and exacerbated neuronal damage17). The expression levels of MSR1 in infiltrating macrophages increase from day 1 to day 3 after stroke onset, which is induced by Mafb, an important transcription factor for the differentiation of macrophages. Macrophages that strongly express MSR1 in the ischemic brain are not major producers of inflammatory cytokines (TNF-α, IL-1β, and IL-23), but instead produce neurotrophic factors, such as insulin-like growth factor1 (IGF-1)17). A recent study demonstrated that post-ischemic treatment by a phthalide derivative that increased the expression of MSR1 in infiltrating macrophages attenuated the production of inflammatory cytokines such as TNF-a through the acceleration of DAMP removal and ischemic neuronal damage in a rat model of ischemic stroke18). Therefore, DAMP removal from ischemic brain tissue is an essential process in ischemic stroke pathologies that determines the functional prognosis.
Macrophages infiltrate ischemic brain tissue and trigger post-ischemic inflammation during the first several days after stroke onset. The number of infiltrating macrophages decreases thereafter (though they reside in ischemic brain tissue for at least 2 weeks), and their phenotype is shifted to anti-inflammatory or reparative19). Infiltrating macrophages are important for long-term recovery, given that CCR2-deficient mice in which macrophage infiltration in ischemic brain tissue is attenuated exhibit exacerbated neurological deficits and functional prognosis from 5 to 28 days after stroke onset20). Some growth factors, such as IGF-1 and FGF-2, are also produced by infiltrating macrophages during the recovery phase of ischemic brain injury, which improve the neurological outcome to protect neurons and glial cells from cell death21,22). FGF-2 also promotes remyelination, and synaptogenesis after ischemic stroke23). Thus, infiltrating macrophages turn their phenotype into anti-inflammatory (pro-resolving) and reparative ones to accelerate functional recovery after ischemic stroke.
Therapeutic treatment methods for ischemic stroke
The intravenous administration of recombinant tissue plasminogen activator (rt-PA) or endovascular thrombectomy immediately after stroke onset is the established effective therapeutic treatment method for ischemic stroke24). Immunosuppressive agents, such as steroids, cyclosporine, and tacrolimus, fail to improve the functional prognosis of human stroke patients. As such, the simple suppression of post-ischemic inflammation is not suitable as a therapeutic strategy against ischemic stroke. Whether neural repair is accelerated or enhanced by immunosuppressive therapeutic methods must be taken into consideration.
Although the anti-inflammatory effects of IL-10 and TGF-β are promising therapeutic targets that are very important for recovery after ischemic stroke, it might be difficult to control the expression of just anti-inflammatory molecules at the appropriate time in the sequential processes of ischemic stroke pathologies25). Recent studies have shown that the transformation of the macrophage phenotype to the anti-inflammatory or pro-reparative one can be observed by the systemic administration of docosahexaenoic acid (DHA), IL-13, and meisoindigo26-28). Treatment by an angiopoietin-1 in a type 1 diabetic rat model of ischemic stroke attenuates the expression of inflammatory factors but promotes vascular and white matter remodeling29). A recent study shows that PPARg and STAT6 may be regulators that can resolve inflammation after ischemic stroke30). Already established therapeutic agents could be used in the anti-inflammation and accelerated recovery treatment of ischemic stroke if the detailed cellular and molecular mechanisms regulating macrophage phenotypes are completely clarified. Although few effective treatments can be administered over 24 h after ischemic stroke onset, it is likely that therapeutics that sustain neural repair in the ischemic brain even if administered a few days after stroke onset can be developed.
Conclusion
Macrophages have inflammatory or anti-inflammatory/pro-reparative functions in ischemic stroke and play different roles in the sequential progression of ischemic stroke pathologies(Fig. 2). Clarification of the detailed mechanisms of macrophage-mediated cerebral inflammation and the subsequent neural repair is key in the development of next-generation therapies for ischemic stroke.
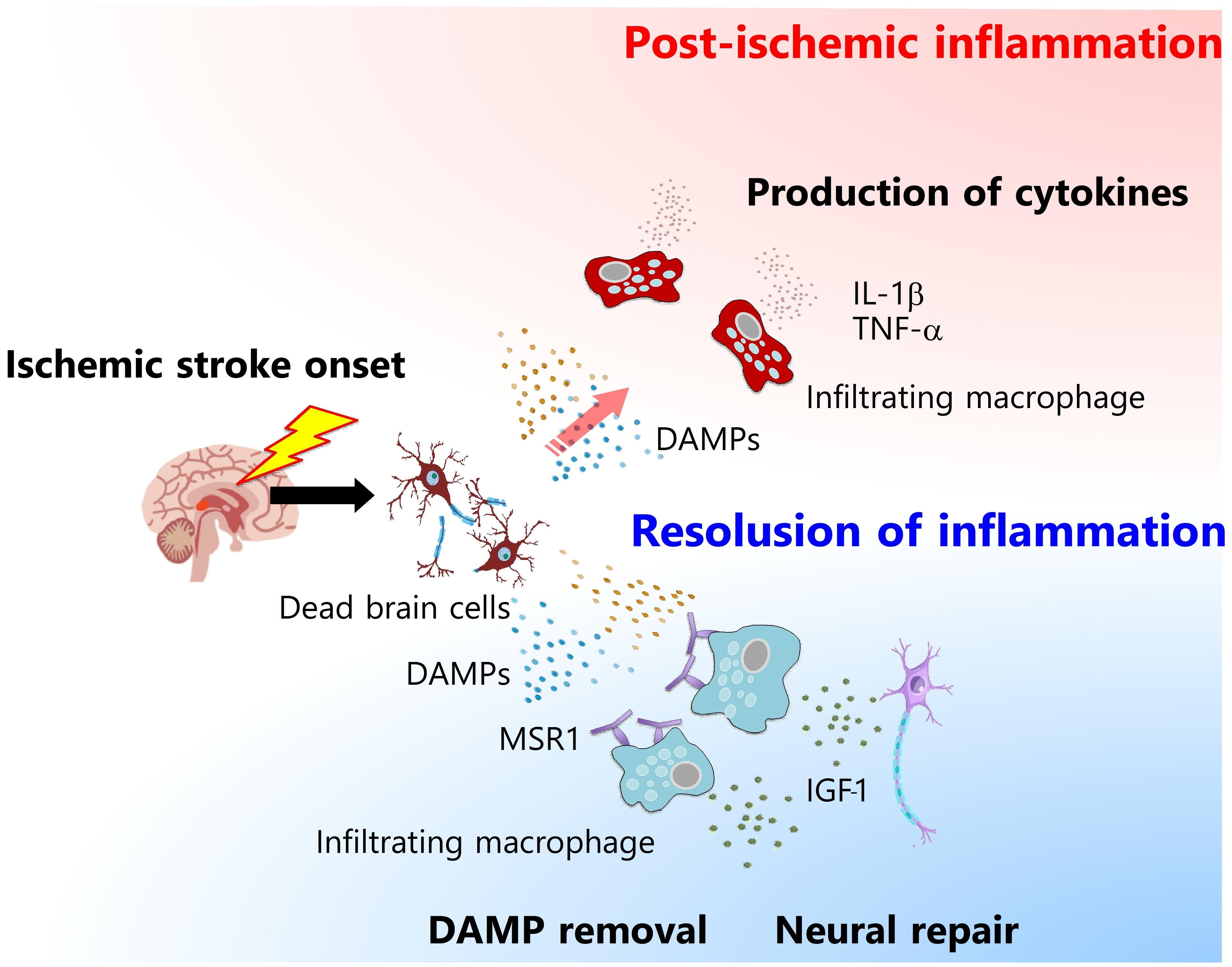 Figure 2: Different phenotypes of infiltrating macrophages in ischemic stroke
Figure 2: Different phenotypes of infiltrating macrophages in ischemic stroke
Abbreviations
BBB: Blood-brain barrier; CCR2: C-C chemokine receptor 2; CCL2: C-C motif chemokine 2; MCP1: Monocyte chemoattractant protein 1; DAMPs: Damage-associated molecular patterns; PRRs: Pattern recognition receptors; HMGB1: High mobility group box1; PRX: Peroxiredoxin; MMP9: Matrix metalloproteinase 9; NETs: Neutrophil extracellular traps; TLRs: Toll-like receptors; IL-1β: Interleukin-1β; TNF: Tumor necrosis factor; TGF-β: Transforming growth factor b; IGF-1: Insulin-like growth factor1; rt-PA: Recombinant tissue plasminogen activator; DHA: Docosahexaenoic acid
Competing interests
The authors declare that they have no competing interests.
References
- Mildner, A. et al. : Nat. Neurosci., 10, 1544 (2007).
- Pedragosa, J. et al. : J. Cereb. Blood Flow Metab., 40, S98 (2020).
- Huang, J. et al. : Ageing Res. Rev., 24, 3 (2015).
- Chin, Y. et al. : J. Neuroinflammation, 10, 95 (2013).
- Chang, X. et al. : Chem. Biol. Interact., 236, 41 (2015).
- Srivastava, P. et al. : Exp. Neurol., 329, 113308 (2020).
- Qiu, J. et al. : J. Cereb. Blood Flow Metab., 28, 927 (2008).
- Zhang, J. et al. : Stroke, 42, 1420 (2011).
- Qiu, J. et al. : Stroke, 41, 2077 (2010).
- Kim, S. W. et al. : Acta Neuropathol. Commun., 7, 94 (2019).
- Rashidian, J. et al. : J. Neurosci., 29, 12497 (2009).
- Shichita, T. et al. : Nat. Med., 15, 946 (2009).
- Shichita, T. et al. : Nat. Med., 18, 911 (2012).
- Clausen, B. H. et al. : Neuroscience, 132, 879 (2005).
- Ooboshi, H. et al. : Circulation, 111, 913 (2005).
- Cekanaviciute, E. et al. : Glia, 62, 1227 (2014).
- Shichita, T. et al. : Nat. Med., 23, 723 (2017).
- Zou, X. et al. : J. Neuroimmune Pharmacol., (2020). doi:10.1007/s11481-020-09911-0.
- Wattananit, S. et al. : J. Neurosci., 36, 4182 (2016).
- Fang, W. et al. : Theranostics, 8, 3530 (2018).
- Ikeda, N. et al. : Stroke, 36, 2725 (2005).
- Zhu, W. et al. : Stroke, 39, 1254 (2008).
- Leker, R. R. et al. : Stroke, 38, 153 (2007).
- Hankey, G. J. : Lancet, 389, 641 (2017).
- Pál, G. et al. : PLoS One, 7, e46731 (2012).
- Ye, Y. et al. : Front. Cell. Neurosci., 13, 553 (2019).
- Cai, W. et al. : Transl. Stroke Res., 9, 669 (2018).
- Kolosowska, N. et al. : Neurotherapeutics, 16, 1304 (2019).
- Venkat, P. et al. : CNS Neurosci. Ther., 27, 48 (2020).
- Zhang, W. et al. : CNS Neurosci. Ther., 25, 1329 (2019).