Vol.7 Amyotrophic lateral sclerosis (ALS)
This article was written by Dr. Seiji Watanabe and Dr. Koji Yamanaka, Department of Neuroscience and Pathobiology, Research Institute of Environmental Medicine, Nagoya University.
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease characterized by upper motor neuron degeneration in the motor area of the cerebral cortex as well as lower motor neuron degeneration in the brainstem and the spinal cord, with the main symptom being systemic muscular atrophy. In Japan, ALS is designated as a specific intractable disease in 1974, with an estimated prevalence of 7 to 10 cases/100,000 persons and an incidence rate of approximately 2 cases/100,000 persons/year.1) This article primarily outlines the molecular pathogenesis of ALS in neurons as well as currently available drugs and biomarkers based on the pathogenesis.
Clinical and pathological feature of ALS
ALS patients present with pathologic reflex and spasticity due to upper motor neuron disturbance, and progressive muscular weakness and atrophy due to lower motor neuron disturbance. As the disease progresses, patients experience gait disturbance, reduced vocal function, and dysphagia, and then finally respiratory failure due to respiratory muscle paralysis. In general, none of sensory disturbance, eye movement impairment, bladder and rectal disturbance, and bedsore, which are collectively known as four major negative clinical signs of ALS, appear. In addition, it is reported that frontotemporal dementia (FTD) involving character change, language disturbance, and impaired cognitive function occurs concurrently in nearly 30% of ALS patients.
Pathologically, specific degeneration and loss of upper and lower motor neurons are observed. Particularly, myelin sheath destruction and abnormal activation of glial cells are notable in the spinal cord. In 2006, Arai et al.2) and Neumann et al.3) reported that TAR DNA binding protein-43 (TDP-43) is the main component of ubiquitin-positive inclusion bodies, which accumulate in neurons in ALS and part of FTD. This discovery indicates that ALS and at least part of FTD may be a group of connected diseases characterized by accumulation of TDP-43 and are based on the common mechanism of disease with abnormal TDP-43 (ALS-FTD spectrum) (Fig. 1).
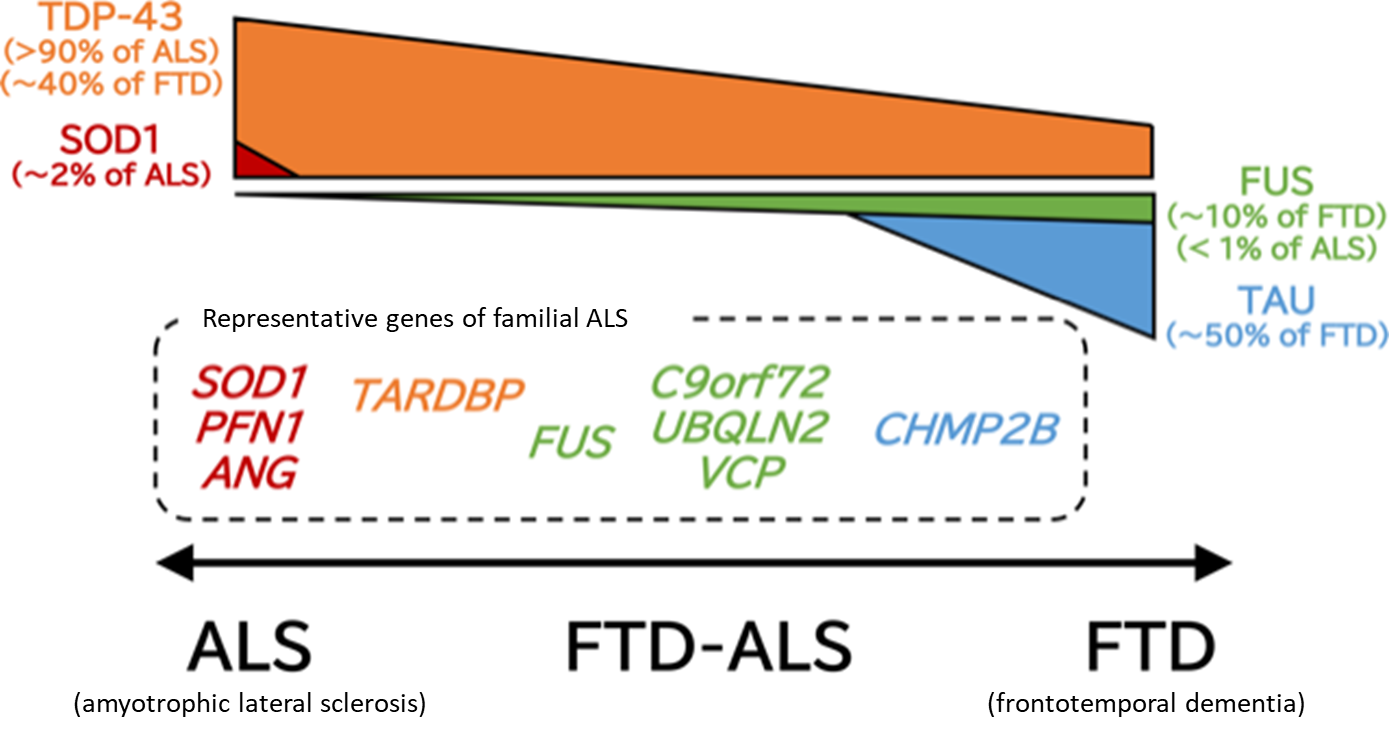
Figure 1. ALS-FTD spectrum
ALS and FTD (frontotemporal dementia) constitute a disease spectrum with the common pathology characterized by accumulation of TDP-43, and FTD-ALS, ALS with higher brain dysfunction, is often observed. This figure presents proteins that accumulate abnormally in the upper part and genes responsible for phenotypically close familial ALS in the lower part according to the ALS-FTD spectrum.
Genetics and animal models of ALS
While more than 90% of ALS cases are sporadic, in other words, not inherited, some are familial ALS, for which more than 20 responsible genes have been identified to date. In Japan, the most common responsible gene is the SOD1 gene, which accounts for approximately 20% of familial ALS. In the West, on the other hand, FTD-ALS with excessively long hexanucleotide repeats in the intron of the C9orf72 gene is predominant, accounting for approximately 30% to 50% of familial ALS. The TARDBP gene, which encodes TDP-43, is another gene responsible for familial ALS, indicating that accumulation of TDP-43 is not secondary to neuronal pathological changes, but is directly involved in the molecular pathogenesis of ALS.
Mutant SOD1 mice engineered to overexpress an SOD1 gene with ALS patient-derived mutation, which present with progressive hind limb paralysis and muscular atrophy due to motor neuron degeneration to reproduce the symptoms and pathology of ALS reliably, have been widely used as an animal model of ALS.4) However, differences from the pathogenesis of sporadic ALS, including the absence of accumulation of TDP-43-positive inclusion bodies, have been pointed out in the mutant SOD1 mice. In recent years, therefore, many attempts are being made to create animal models that mimic sporadic ALS, which is overwhelmingly predominant in ALS. While mice expressing ALS-derived mutant TDP-435) and transgenic mice carrying abnormal hexanucleotide repeat expansion in intron in the C9orf72 gene6) have been reported, model animals reproducing motor neuron-specific cell death have not been established, leaving an important issue.
Pathophysiology of ALS
To date, various possible mechanisms underlying the molecular pathogenesis of ALS have been reported, including excitotoxicity, oxidative stress, mitochondrial damage, endoplasmic reticulum stress, collapse of the proteolytic system, and abnormal RNA metabolism (Table 1). Of note, edaravone (brand name: RADICUT) was recently approved for inhibition of progressive functional impairment in ALS in association with oxidative stress, and perampanel (brand name: FYCOMPA), an antiepileptic drug, was shown to be protective against excitotoxicity in an animal model.7) Since TDP-43 aggregates are extensively observed in ALS, there is no doubt about the pathogenetic involvement of TDP-43, but its related molecular mechanism is under discussion. Dominant inheritance of mutant TDP-43-mediated ALS and motor dysfunction in mice overexpressing TDP-43 suggest a gain-of-toxicity hypothesis. On the other hand, embryonic lethality of TDP-43-deficient mice, neuronal death resulting from deletion of TDP-43 in adult mice, and loss of TDP-43 from the nucleus, where TDP-43 is normally localized, in ALS indicate neuronal degeneration due to loss of function of TDP-43, requiring further study to elucidate the mechanism. Recently, multiple groups, including us, reported functional impairment of mitochondria-associated membrane (MAM) in ALS8,9) (Fig. 2). The MAM, a subdomain of the endoplasmic reticulum membrane that is in close contact with the outer mitochondrial membrane, is a microdomain known to have various physiological functions, including Ca2+ supply to the mitochondria, innate immunity, and control of autophagy. Collapse of MAM may be expected to explain the impairment of two different organelles, that is, mitochondria and endoplasmic reticulum, in ALS consistently.
The prion-like spread of TDP-43, like α-synuclein in Parkinson's disease and tau in Alzheimer's disease, in the form of pathological aggregates via β-sheet structure is proposed.10) More specifically, abnormal TDP-43 aggregates in the cytoplasm may alter the structure of normal TDP-43 to take up TDP-43 inside; in fact, it is known that normal TDP-43 is lost from the neuronal nucleus, where TDP-43 is normally localized, in ALS patients with TDP-43-positive inclusion bodies in the cytoplasm. It was recently shown that these aggregates are formed by so-called liquid-liquid phase separation11,12,13) (Fig. 3). Liquid-liquid phase separation is especially seen in RNA-binding proteins, and these proteins may physiologically control RNA metabolism by constructing small membrane-free organelles in the cytoplasm through self-association. While aggregation and dispersion via liquid-liquid phase separation are usually reversible, various factors such as environmental stress may make aggregation irreversible, resulting in abnormal aggregate formation. In fact, multiple genes (e.g., HNRNPA1, FUS, TIA1) for RNA-binding proteins with liquid-liquid phase separation, in addition to TDP-43, are known genes responsible for familial ALS, indicating that abnormal liquid-liquid phase separation may be the molecular pathogenesis underlying abnormal RNA metabolism in ALS.
Table 1. Hypothesis for the pathogenesis in neurons in ALS
Pathogenetic mechanisms proposed to date and representative relevant ALS-related genes are presented.
| Pathogenetic hypothesis | Representative relevant ALS-related gene | |
|---|---|---|
| Excitotoxicity | Overactivation of motor neurons Excessive Ca2+ release into the cytoplasm |
SOD1, ADARB1, TARDBP |
| Impaired proteolysis | Accumulation of abnormal proteins Proteasome impairment Endoplasmic reticulum stress |
SOD1, TARDBP, FUS, SQSTM1, TBK1, VAPB, VCP |
| Mitochondrial impairment | Increased reactive oxygen species Reduced ATP synthesis |
SOD1, TARDBP, VAPB, SIGMAR1 |
| Abnormal RNA metabolism | RNA aggregate formation Failure of splicing control |
TARDBP, FUS, C9orf72, HNRNP1 |
| Impaired intracellular transport | Impaired axonal transport Abnormal cytoskeleton |
SOD1, DCTN1, PFN1, TUBA4A |
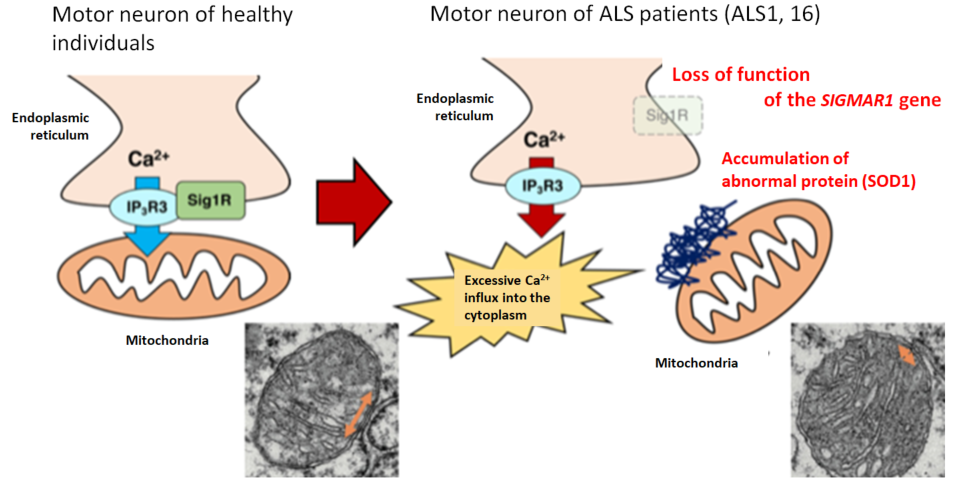
Figure 2. Collapse of mitochondria-associated membrane (MAM) in ALS
The MAM (orange arrow) was reduced in mice with genetic abnormality responsible for familial ALS compared with wild-type mice, indicating that collapse of MAM may promote neuronal degeneration through abnormal Ca2+ control in ALS (modified from EMBO Mol. Med. by Watanabe et al. (2016)).
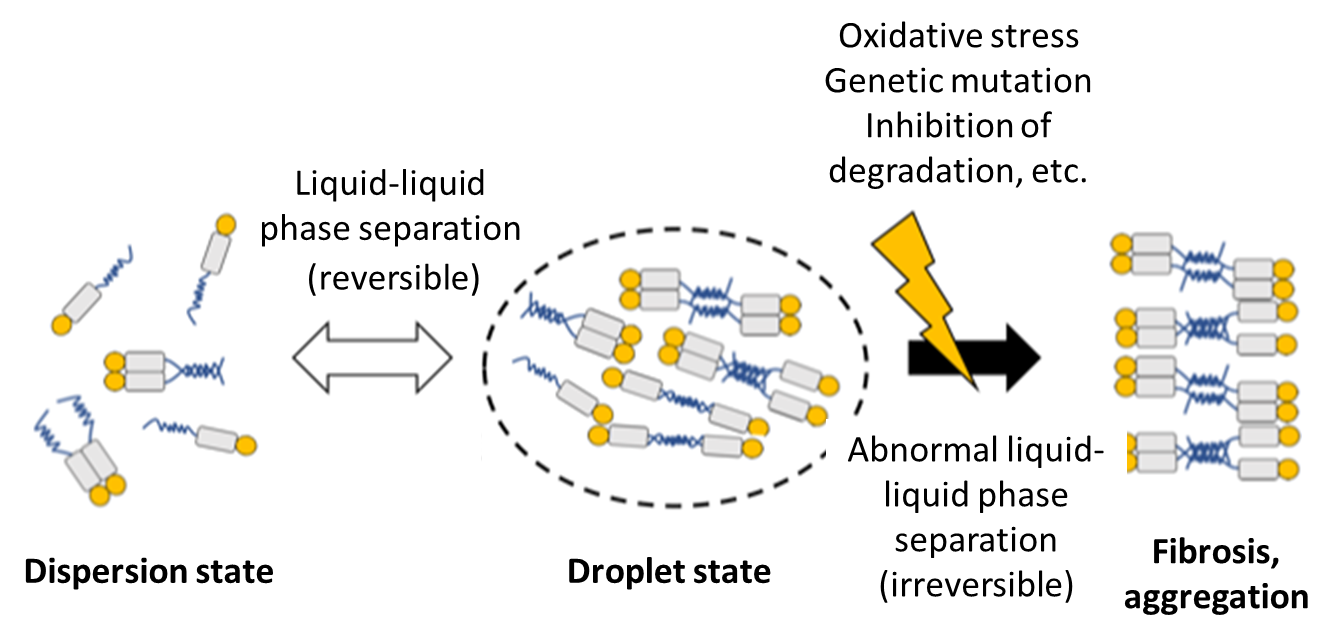
Figure 3. Hypothesis for TDP-43 aggregation through liquid-liquid phase separation
RNA-binding proteins, including TDP-43, have structurally unstable region(s) (blue line), and these regions mutually interact to assemble in a droplet state. This phase separation is usually reversible and physiological, but may become abnormal due to external stress or other factors, resulting in aggregation or inclusion body formation.
Non-cell autonomous neuronal death
A marked increase in reactive gliosis such as microglia and astrocyte activation in the spinal cord has been observed in ALS patients and animal models. While this change was previously considered secondary to motor neuron degeneration, glial cell-specific deletion of mutant SOD1 prolonged the survival of mutant SOD1 mice, indicating that glial cells are actively involved in the pathogenesis of ALS to control motor neuron degeneration.14,15) Neuronal death caused by abnormal glial cells, which are non-neuronal cells, in this manner is called non-cell autonomous neuronal death. In recent years, the pathogenesis of ALS is being analyzed with focus on abnormal glial cells, revealing the involvement of molecules in glial cells such as the cytokine TGF-β16) and the intracellular transcription factor NFκB17) in activation of glial cells and exacerbation of disease. Since glial cells, which may define disease progression, are an important target for disease-modifying therapy after the onset of ALS, methods for controlling the activation of glial cells should be developed based on the latest findings.
Disease-modifying therapy for ALS
Unfortunately, no curative treatment is yet available for ALS. However, new drugs are being developed for disease-modifying therapy to slow disease progression based on the pathogenesis and improve the survival and quality of life (QOL) of patients. Riluzole (brand name: RILUTEK) was first approved for the treatment of ALS in 1996 and is still widely used as first-line treatment. More recently, edaravone was approved in Japan. Edaravone may delay disease progression by suppressing oxidative stress, which increases in ALS patients and accelerates motor neuron degeneration. In addition, various clinical studies are currently underway in Japan and the rest of the world, including those of high-dose methylcobalamin and intrathecal hepatocyte growth factor.18) Since ALS progresses very rapidly after the onset and motor neurons may have already been considerably damaged at the clinical onset, it is desirable to start treatment as early as possible. In addition, disease progression and prognosis greatly vary among individuals: while approximately 10% of patients die within 1 year after the onset, at least 10-year survivors have been reported, although most patients survive for several years.1) At present, no reliable biomarker for early diagnosis of ALS or predicting the prognosis of individual patients has yet been established, requiring further study to develop useful biomarkers.
Conclusion
In recent years, the pathogenesis of neurodegenerative diseases, including ALS, has been increasingly elucidated, producing many findings. However, only a few drugs have been shown to inhibit the progression of ALS with very limited efficacy. The aforementioned deficiency of animal models and biomarkers as well as the pathogenetic differences between familial ALS and sporadic ALS may be largely responsible. These issues should be resolved through research based on new technologies, including iPS cells and genome editing, to develop more effective treatments.
References
- 辻 省次、祖父江 元:「すべてがわかるALS・運動ニューロン疾患(アクチュアル脳・神経疾患の臨床)」(中山書店)(2013)
- Arai, T. et al . : Biochem. Biophys. Res. Commun ., 351, 602-611(2006)
- Neumann, M. et al . : Science , 314, 130-133(2006)
- Gurney, M. E. et al . : Science , 264, 1772-1775(1994)
- McGoldrick, P. et al . : Biochim. Biophys.Acta , 1832, 1421-1436(2013)
- Chew, J. et al . : Science , 348, 1151-1154(2015)
- Akamatsu, M. et al . : Sci. Rep ., 6, 28649(2016)
- Stoica, R. et al . : Nat. Commun. , 5, 3996(2014)
- Watanabe, S. et al . : EMBO Mol. Med ., 8, 1421-1437(2016)
- Nonaka, T. et al . : Cell Rep ., 4, 124-134(2013)
- Conicella, A. E. et al . : Structure , 24, 1537-1549(2016)
- Murakami, T. et al . : Neuron , 88, 678-690(2015)
- Patel, A. et al . : Cell , 162, 1066-1077(2015)
- Yamanaka, K. et al . : Nat. Neurosci ., 11, 251-253(2008)
- Boillee, S. et al . : Science , 312, 1389-1392(2006)
- Endo, F. et al . : Cell Rep ., 11, 592-604(2015)
- Ouali Alami, N. et al . : EMBO J ., e98697(2018)
- 岸川忠彦:「今動いている臨床研究・試験と治験情報」,日本ALS 協会会報, 101, 16-17(2017)