2nd review: Origin of microglia and brain diseases
This article was written by Dr. Kazuyuki Takata, Division of Integrated Pharmaceutical Sciences, Kyoto Pharmaceutical University.

Introduction
Microglia are tissue macrophages in the central nervous system (CNS) (i.e., brain and spinal cord) and play a frontline role in CNS immunity. With its unique developmental mode and biology being revealed, microglia have drawn much attention from scientists in various academic fields, resulting in further extensive biological research of microglia. With focus on the developmental origin and biology of microglia, this article introduces their involvement in brain diseases and validity as a treatment target.
Ontogeny of microglia
In 1919, Pío del Río Hortega, a Spanish neuroscientist, clearly described the morphological features of cells constituting the brain as the third group following neurons and astrocytes, and called this cell population microglia. Referring to the developmental origin of microglia, Hortega mentioned that their progenitors migrate to the brain during very early developmental stage. Peripheral monocytes had been considered microglial precursors over decades until an experiment in mice identified primitive macrophages arising from hematopoiesis in the extraembryonic yolk sac as their precursor 10 years ago.1) The primitive macrophages develop from the erythro-myeloid progenitors (EMPs) in a transcription factor Myb-independent manner2) without transition into monocytes and then temporarily locate in various tissues throughout the body, including the brain, as tissue macrophages (Figure). Later, EMPs migrate to the fetal liver, a new place of hematopoiesis where EMPs differentiate into monocytes (fetal liver monocytes) in a Myb-dependent fashion. These monocytes circulate throughout the body and form a population as new tissue macrophages by replacing or coexisting with primitive macrophages residing earlier.3) At this time, the blood-brain barrier (BBB) is developing in the brain, practically preventing the invasion of fetal liver monocytes. Thus, while resident tissue macrophages exist in organs throughout the body, only microglia primarily derive from primitive macrophages in the yolk sac.
Characterization of microglia based on cellular origin
Later than hematopoiesis by EMPs, hematopoietic stem cells (HSCs) develop in the embryonic aorta-gonad-mesonephros (AGM) region (Figure). HSCs start embryonic hematopoiesis in the mouse fetal liver, but migrate to the bone marrow immediately before birth to play a general role in definitive hematopoiesis after birth. HSC-derived monocytes may be another source of tissue macrophages, but hardly contributes to the microglial population under physiological conditions.1, 4) On the other hand, HSC-derived monocytes circulate throughout the body via blood flow and migrate to tissues to differentiate into macrophages in response to infection or tissue-injury signals. These HSC monocyte-derived macrophages are short-lived and temporarily reside in tissues to provoke an inflammatory response, whereas EMP-derived tissue-resident macrophages may play a major role in maintenance and tissue remodeling. As evidenced by high expression of the chemokine receptor CCR2 on HSC-derived macrophages and high expression of F4/80, colony-stimulating factor-1 receptor (CSF-1R), and the fractalkine receptor CX3CR1 on EMP-derived macrophages, individual gene expression patterns reflect the cellular origin4) and may result in functional role assignment. CSF-1R-mediated signaling inducing microRNA-21 to inhibit the expression of pro-inflammatory factors and promote the expression of neurotrophic factors5) is interesting to explain the functional characteristics attributable to the cellular origin of EMP-derived tissue macrophages including microglia.6) Recently, it has been suggested that both macrophages in the perivascular space, dura mater, and choroid plexus, which are adjacent to the brain parenchyma (border-associated macrophages: BAMs), and microglia in the parenchyma derive from EMPs in the yolk sac, but they are differently fated when they are still in the yolk sac,7) and gene expression patterns that distinguish BAMs from microglia have been identified.8) Collectively, the features of microglia may reflect not only signals received from the brain or spinal cord environment that they reside in, but also the influence of their unique developmental origin, indicating that the developmental origin should be taken into consideration to understand the biology and function of microglia more deeply and accurately.
Maintenance of the microglial population
As mentioned above, microglial progenitors are produced only during embryonic development, and postnatal bone marrow HSC-derived monocytes are not practically involved in the replenishment of microglia. Concerning postnatal maintenance of the microglial population, it has recently been reported that coupled apoptosis and cell division are repeated in the mouse brain in such a manner that microglia in the whole brain are completely replaced in a cycle of approximately 96 days, maintaining a constant number of microglia in the brain.9) This mechanism for maintaining the population based on self-division may be greatly involved in the appearance of spatio-temporal dependent microglial subpopulations and disease-specific subpopulations by transmitting information of developmental origin and stimuli from constantly changing microenvironment to daughter cells in the form of epigenetic modification.10, 11)
Microglia and brain diseases
Down syndrome (DS), which is due to trisomy 21, is associated with reduced embryonic neurogenesis of unknown mechanism. A collaborative study with Dr. Keiichi Ishihara of Kyoto Pharmaceutical University using DS model mice revealed reduced proportion of brain macrophages, microglia and BAMs, and increased proportion of inflammatory cells such as neutrophils and monocytes, as well as reduced neurogenesis in the cerebral cortex in the embryonic brain12) (Figure). These results suggest that a dysregulated immune environment in the embryonic brain may be deeply involved in the development of DS and that the improvement by in utero treatment may be a novel therapeutic strategy for DS.
Meanwhile, the greatest risk factor for Alzheimer's disease (AD) is aging, and accumulation of amyloid-β (Aβ) in the brain over decades before the onset of clinical symptoms is recognized as the etiological trigger. In addition, it is conventionally well known that microglia accumulate at sites of Aβ deposition (senile plaque). Considering the mechanism for maintaining the microglial population based on self-proliferation, the microglial dysregulation over time due to aging and long-term exposure to Aβ13) may be a primarily risk factor connecting between Aβ accumulation and development of AD. In AD model mice with Aβ accumulation in the brain, we found that cognitive dysfunction was improved by hippocampal transplantation with macrophages which were prepared from mouse bone marrow- or peripheral blood-derived HSCs by CSF-1 stimulation14, 15) (Figure). It was found that the therapeutic effect was due not only to Aβ phagocytosis by transplanted macrophages, but also to resident microglial functional changes caused by transforming growth factor-β1 (TGF-β1) secreted from the transplanted cells.16) TGF-β signaling is reported to be essential to destine primitive macrophages to become microglia during early development,7) whereas clearance of aged glial cells, including microglia, is reported to result in improved cognitive function.17) Microglial dysregulation over time due to aging and long-term exposure to Aβ may result in cognitive deficit, and TGF-β signaling, which is essential for microglial maturation in an early stage of developmental, may partially attenuate the dysregulation to improve cognitive function.
Conclusion
Through the mechanism for maintaining the microglial population by repeating self-proliferation, the information of developmental origin and stimuli from the microenvironment may be transmitted in the form of epigenetic modification to characterize microglia in a spatio-temporal dependent manner. In a collaborative study with Dr. Florent Ginhoux of Agency for Science, Technology and Research (A*STAR), we successfully generated primitive macrophage (iMacs) reflecting embryonic hematopoiesis from induced pluripotent stem (iPS) cells (Figure).18) Study using such primitive cells that can reproduce the developmental origin is expected to help to understand the biology of microglia more accurately. To clarify pathogenetic mechanisms and obtain clues for the treatments against brain diseases arising in various stages from development to aging, it may be very important to understand the microglial program from ontogeny to maturation and even aging accurately.
The macrophage transplantation study in AD model mice was conducted in collaboration with Professor Shun Shimohama of Sapporo Medical University, Professor Eishi Ashihara of Kyoto Pharmaceutical University, and Professor Yoshihisa Kitamura of Ritsumeikan University.
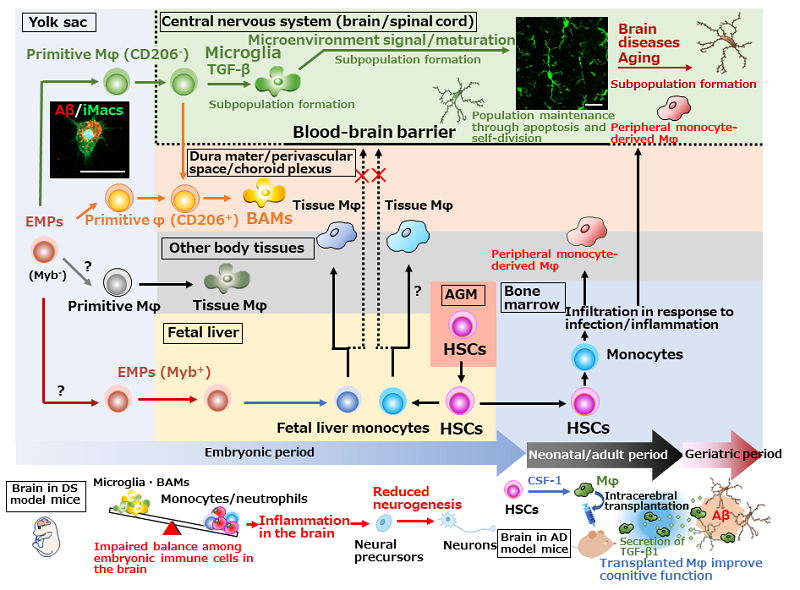
Figure. Ontogeny of tissue macrophages, including microglia, and brain diseases
Primitive Mφ are produced from EMPs in the mouse yolk sac in a transcription factor Myb-independent manner without transition of monocytes and then locate in various tissues as tissue Mφ, including microglia. Primitive Mφ are already destined to become microglia or BAMs at this time and require TGF-β signaling to differentiate into microglia. It is unclear how the other primitive Mφ are destined. Later, EMPs migrate to the fetal liver to produce fetal liver monocytes, which then migrate and differentiate into new tissue Mφ population by replacing or coexisting with primitive Mφ. CNS primitive Mφ, which are protected by the BBB, are not replaced by fetal live monocytes. HSCs originally arose in AGM region also produce monocytes, but the HSC-derived monocytes basically do not make up microglial population under physiological conditions, but migrate from blood to tissues to differentiate into Mφ in response to infection/tissue-injury signals. Microglia develop from primitive Mφ in the yolk sac and maintain the population through coupled apoptosis and self-proliferation. In DS model mice, reduced proportion of microglia and BAMs, increased proportion of inflammatory cells, and reduced neurogenesis are observed in the embryonic brain.12) In AD model mice, cognitive dysfunction is improved after the transplantation with Mφ prepared from HSCs by CSF-1 stimulation. Therapeutic mechanisms are not only Aβ phagocytosis by transplanted cells, but also Aβ phagocytosis by resident microglia which are promoted by TGF-β1 secreted from transplanted cells.14-16) Left photo: iPS cell-derived Mφ (iMacs) reproduce the developmental process of primitive Mφ and can be differentiate into microglia which have Aβ phagocytic activity. Right photo: Microglia in the adult mouse hippocampus (stained with goat anti-Iba1 antibody)
Scale bars: 20 μm. Aβ: amyloid-β, AD: Alzheimer's disease, AGM: aorta-gonad-mesonephros, BAMs: border-associated macrophages, CSF-1: colony-stimulating factor-1, DS: Down syndrome, EMPs: erythro-myeloid progenitors, HSCs: hematopoietic stem cells, Mφ: macrophages, TGF-β: transforming growth factor-β.
References
- Ginhoux, F. et al. : Science, 330, 841 (2010).
- Schulz, C. et al. : Science, 336, 86 (2012).
- Hoeffel, G. et al. : Immunity, 42, 665 (2015).
- Hagemeyer, N. et al. : EMBO J., 35, 1730 (2016).
- Caescu, C. I. et al. : Blood, 125, e1 (2015).
- Chitu, V. and Stanley, E. R. : Curr. Top. Dev. Biol., 123, 229 (2017).
- Utz, S. G. et al. : Cell, 181, 557 (2020).
- Mrdjen, D. et al. : Immunity, 48, 380 (2018).
- Askew, K. et al. : Cell Rep., 18, 391 (2017).
- Masuda, T. et al. : Nature, 566, 388 (2019).
- Keren-Shaul, H. et al. : Cell, 169, 1276 (2017).
- Ishihara, K. et al. : Brain Pathol., 30, 75 (2020).
- Baik, S. H. et al. : Cell Metab., 30, 493 (2019).
- Kawanishi, S. et al. : J. Alzheimers Dis., 64, 563 (2018).
- Kuroda, E. et al. : J. Alzheimers Dis., 73, 413 (2020).
- Kuroda, E. et al. : Neuroscience, 438, 217 (2020).
- Tyler, B. et al. : Nature, 502, 578 (2018).
- Takata, K. et al. : Immunity, 47, 183 (2017).