Vol.5 Overview of Parkinson's Disease
This article was written by Dr. Hodaka Yamakado, Department of Neurology, Graduate School of Medicine, Kyoto University
Introduction
Parkinson's disease (PD) is a neurodegenerative disease clinically characterized by Parkinson symptoms and pathologically characterized by Lewy bodies and dopaminergic neuronal death. PD is the most common neurodegenerative disease second to Alzheimer's disease, with a prevalence of 100−200 cases per 100,000 persons in developed countries. While various possible mechanisms underlying the development of PD have been proposed from genetic studies, concepts of PD as a prion disease have emerged based on pathological studies by Braak et al. and biochemical properties of α-Synuclein, the main component of Lewy bodies. This article will review the pathophysiology of PD as well as the recent progress of the development of drugs and early biomarkers.
Clinical and pathological picture of PD
Motor symptoms of PD include tremor, rigidity, akinesia, and postural instability, which are known as four major symptoms of PD, but many patients do not present with akinesia or postural instability in the early stage. In addition, non-motor symptoms, including reduced sense of smell, REM sleep behavior disorder (RBD), constipation, frequent urination, and orthostatic hypotension, often occur concurrently with or sometimes precede the motor symptoms.
Pathologically, inclusion bodies known as Lewy bodies are observed in neurons, with the main component being α-Synuclein (α-Syn) aggregates (Fig. 1). Findings from consecutive brain biopsy cases suggest that Lewy bodies first occur in the dorsal motor nucleus of vagal nerves, olfactory bulb, or peripheral autonomic nerve, and in the brainstem spread upward to the locus coeruleus, raphe nucleus, and substantia nigra, and further to the cerebral cortex1) (Fig. 2). Corresponding symptoms include constipation, frequent urination, and orthostatic hypotension due to the dysfunction of peripheral autonomic nervous system, and non-motor symptoms such as depression and sleep disturbance due to the damaged locus coeruleus and raphe nucleus.2) Eventually, dysfunction of nigral dopaminergic neurons manifests as Parkinson's motor symptoms, and dysfunction of cerebral cortex terminally manifests as dementia. There is a moderate correlation between α-Syn accumulation and cell death. In summary, Lewy bodies and cell death in specific cell groups are pathological characteristics of PD, especially in the early stage.
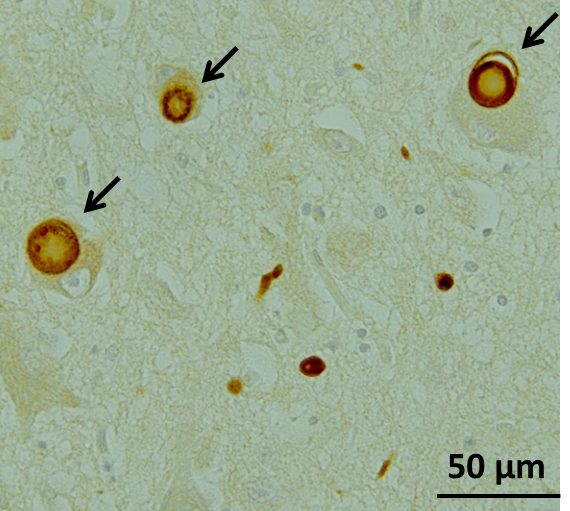
Figure1. Lewy bodies in the substantia nigra in a patient with PD, stained with anti-phosphorylated α-Syn antibody (Image courtesy of Dr. Takashi Ayaki, Department of Neurology Kyoto University Graduate School of Medicine)
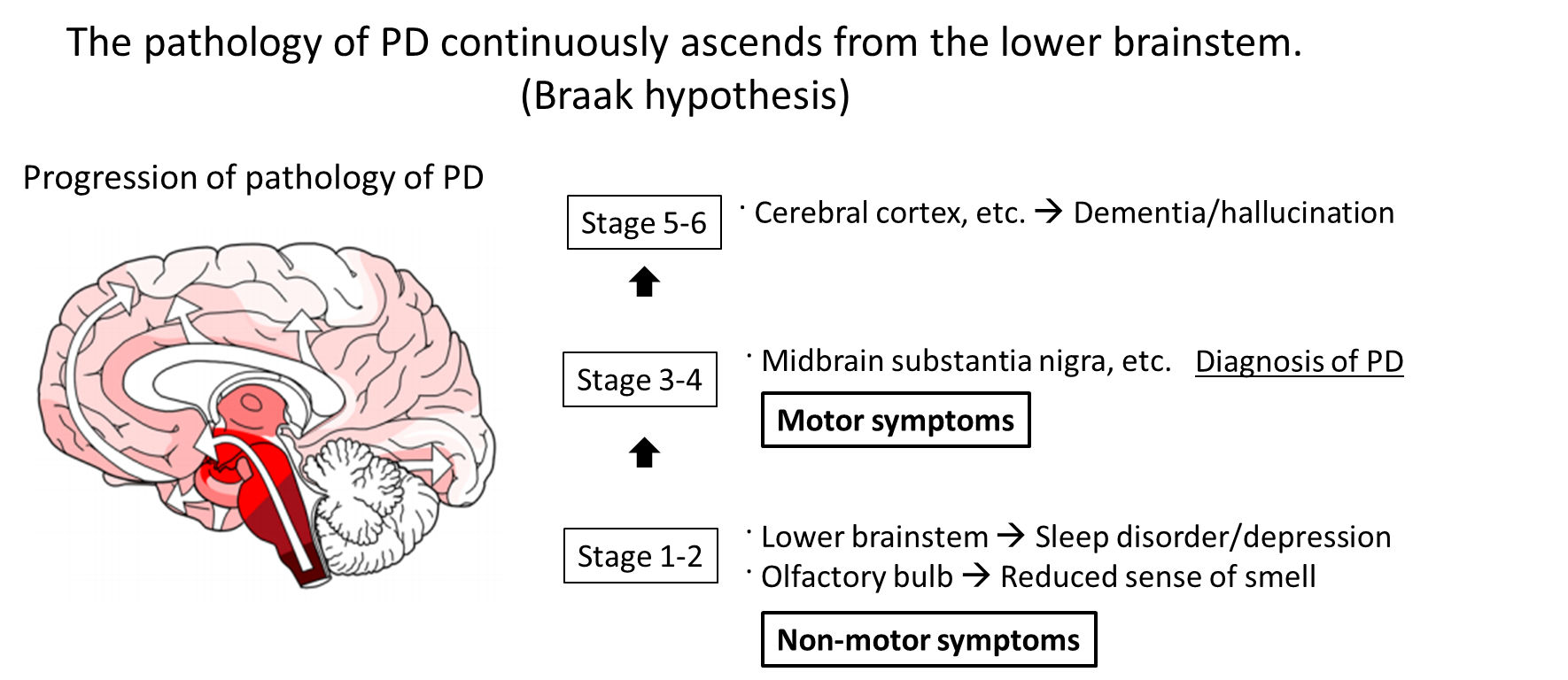
Figure 2. It is assumed that the pathology of PD typically occurs in the olfactory bulb or dorsal motor nucleus of vagal nerves and in the brainstem spreads upward to the locus coeruleus and substantia nigra, and further to the cerebral cortex.18) Corresponding symptoms include non-motor symptoms such as depression and sleep disorder due to damage to the lower brainstem including the locus coeruleus. Eventually, nigral dopaminergic neuron damage manifests as Parkinson symptoms, and cerebral cortex damage terminally manifests as dementia. Twenty years or more may pass in total process.
Pathophysiology of PD
1. Findings in familial PD
Mechanisms underlying the development of PD have been studied mainly based on findings in familial PD since the late 1990s. To date, more than 20 familial PD genes as well as several disease susceptibility genes have been identified. The familial PD genes are classified into several groups according to their roles and functions. One is a group of genes directly related to α-Syn, a component of Lewy bodies: point mutations or multiplications in the α-Syn gene may result in Lewy body formation and cell death due to increased α-Syn aggregation.3,4,5) Besides, there are genes such as parkin involved in the maintenance of mitochondrial function, genes involved in vesicular transport, and genes involved in the proteolytic system. Since some patients with familial PD have Parkinson symptoms and dopaminergic neuronal death without Lewy bodies or with different distribution of Lewy bodies from those of sporadic PD, findings in familial PD should be carefully interpreted when applied to sporadic PD. In this regard, familial PD due to α-Syn gene duplication is clinically and pathologically very similar to sporadic PD,4,5) suggesting the importance of quantitative change of α-Syn in sporadic PD as well.
2. GWAS and rare variants in sporadic PD
Genome-wide association studies (GWAS) in sporadic PD have also revealed important findings. They have identified single nucleotide polymorphisms (SNPs) of α-Syn gene and LRRK2 genes which are responsible for familial PD as risk factors in sporadic PD,6,7) indicating a close involvement of these genes in the development of sporadic PD as well. In addition, MAPT, which encodes tau protein, and HLA, which is involved in immunity, have been identified, but a detailed mechanism has yet to be elucidated. It should be noted that the odds ratio is at most 1.3 for these risk genes. Although high incidence of PD in families with Gaucher's disease (GD), a rare lysosomal storage disease, were already reported, heterozygous mutations in the glucocerebrosidase gene (GBA), whose homozygous mutations are responsible for GD, are also found to be a strong risk factor for PD with an odds ratio of 5.43.8) For example, in Japan these mutations are observed in less than 1% of healthy individuals (thus, they are so-called rare variants), whereas it is reported that approximately 10% of patients with PD have a heterozygous GBA mutation,9) indicating that these mutations are an important risk factor with relatively high frequency and odds ratio (Fig. 3). While a detailed mechanism has yet to be elucidated, accumulation of the substrate for GBA, a lysosomal glycolipid-degrading enzyme, may result in reduced lysosomal function and accumulation of α-Syn.10)
3. Prion hypothesis in PD
Possible mechanisms underlying the development and progression of PD have also been studied based on findings from pathological studies and biochemical analyses of α-Syn. Brain autopsy series showed that accumulation of α-Syn begins in the olfactory bulb or dorsal motor nucleus of vagal nerves, which are externally exposed via the olfactory mucosa and gastrointestinal tract, respectively, and continuously spreads in the brains. These findings are consistent with the idea that α-Syn can be transferred to neighbouring cells and has prion-like properties. More specifically, insoluble α-Syn species with an abnormal secondary structure such as β-sheet structure act as a seed to change the structure of surrounding normal α-Syn successively. Actually, aggregated α-Syn in Lewy bodies has a β-sheet structure, and an experimental study showed that injected synthetic α-Syn polymers with a β-sheet structure spread in the mice brain and acted like a prion.11) This can lead to a hypothesis that some external stimulus-induced aggregation and structural changes of α-Syn in the lower brainstem or olfactory bulb may spread slowly in the brains over 10 years or more. This is the prion hypothesis in PD, and treatments targeting cell-to-cell transmission of α-Syn are being developed based on the hypothesis.
4. Other
It is reported that the localization of Lewy bodies and neuronal death slightly differs in PD. Highly vulnerable neurons with a high incidence of cell death are dopaminergic cells in the substantia nigra and noradrenergic cells in the locus coeruleus. The L-type calcium channels, which are responsible for pacemaker firing of these cells, are reported to be involved in these neuronal deaths. More specifically, the increase in oxidative stress due to a calcium influx via the channel results in cell death,12) with Lewy body formation as a secondary change. In fact, it is reported that the in vitro application of an L-type calcium channel inhibitor reduced oxidative stress without affecting the pacemaker rhythm in dopaminergic neurons.
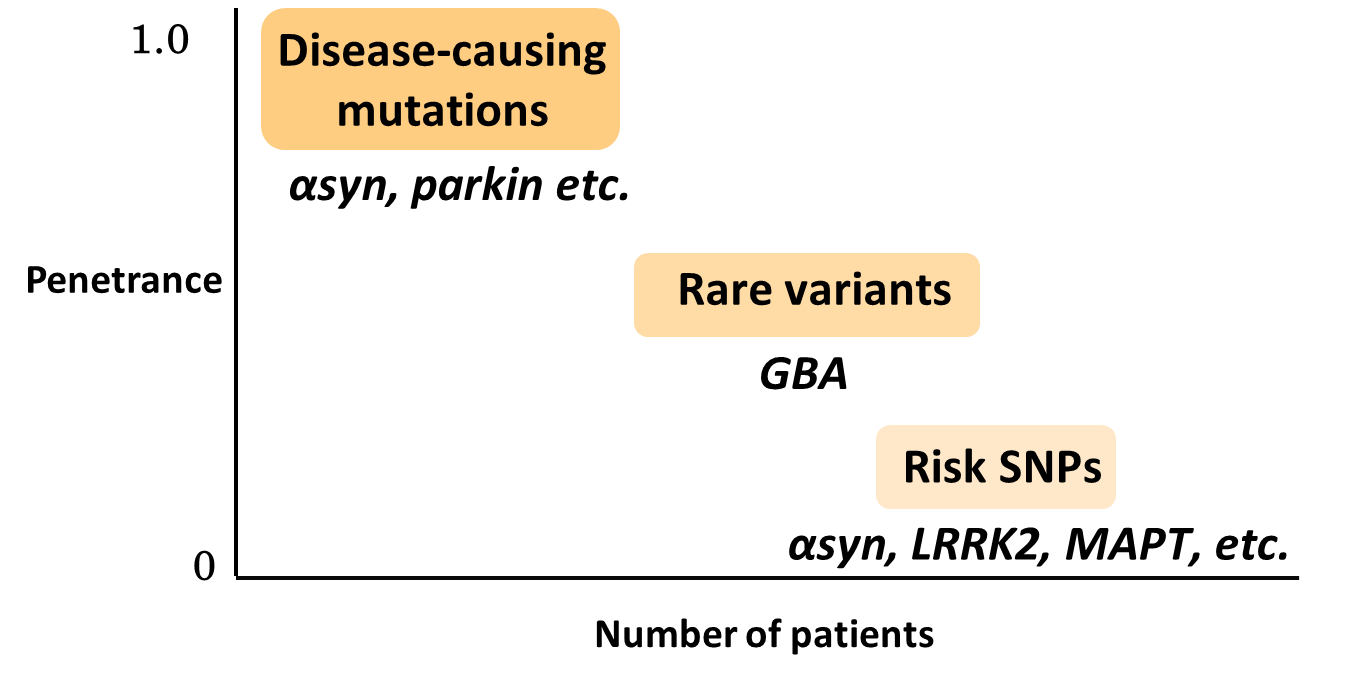
Figure 3. PD-related genes. Gene mutations responsible for familial PD are infrequent but responsible for high penetrance rate. Risk SNPs are frequent but penetrance rate are low. Rare variants are the intermediate.
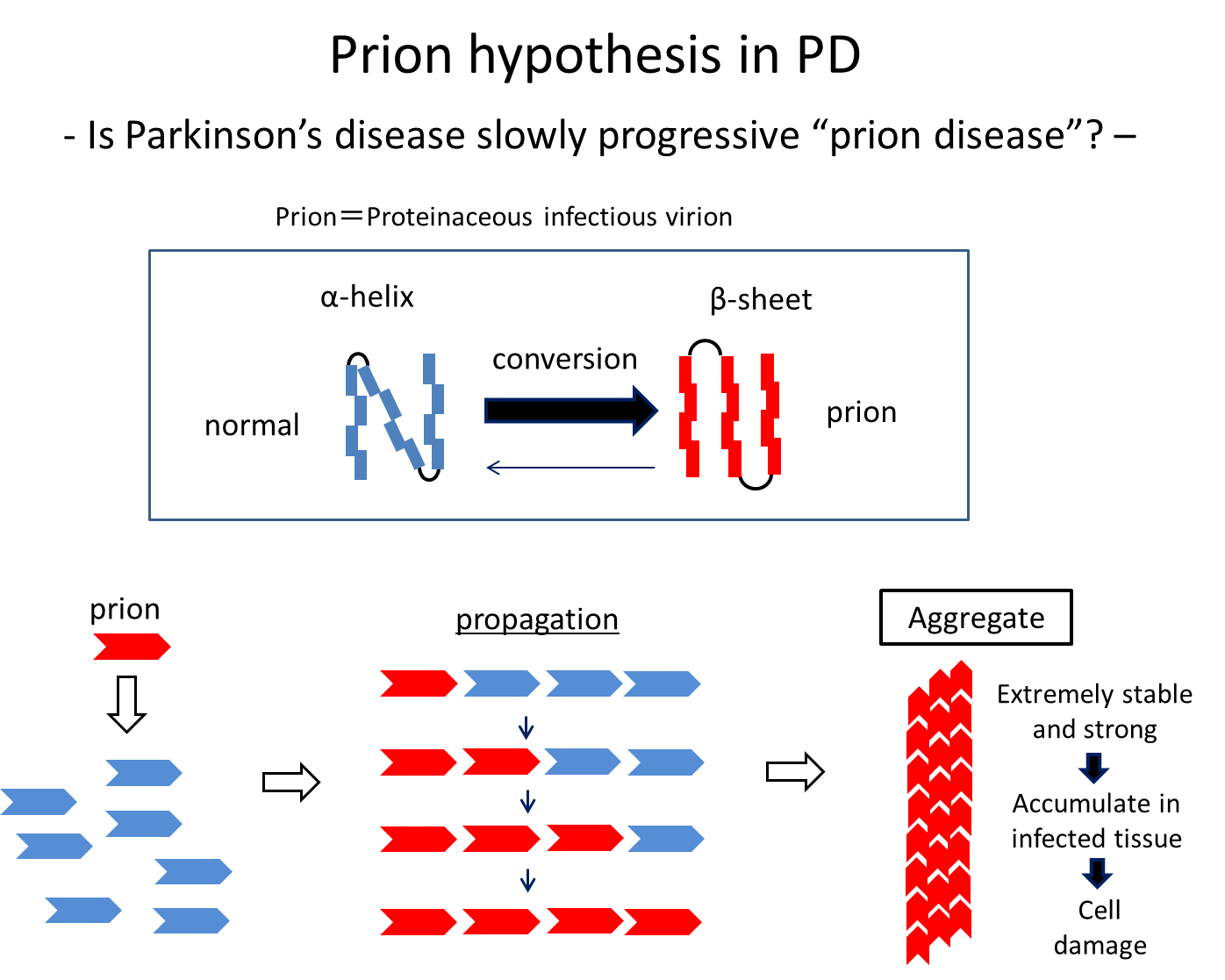
Figure 4. Some stimuli convert normal proteins with an α-helix structure into abnormal proteins with as β-sheet structure, which then spread by abnormalizing the structure of surrounding normal proteins successively. It is known that α-Syn can spread, undergoing these structural changes at least in vitro and in model animals.
Symptomatic therapy and disease-modifying therapy (DMT) for PD
L-dopa has been established as the gold standard treatment for PD. Other anti-PD drugs, including dopamine receptor agonists and inhibitors of dopamine-degrading enzymes (e.g., monoamine oxidase), are also commercially available, but all these drugs are used for symptomatic therapy. It is only in this century that several drugs are being developed for disease-modifying therapy (DMT) to change the natural course of PD (inhibit the progression). All these therapeutic attempts are based on the hypothesis for the PD pathogenesisand are therefore considered proof-of-concept treatment.
Based on the prion hypothesis of α-Syn, anti-α-Syn antibody therapy (passive immunotherapy) with an aim to capture α-Syn at the site of cell-to-cell propagation and α-Syn vaccine therapy (active immunotherapy) are primarily being developed13): the former is currently being tested in a Phase II study,14) and the latter has just completed a Phase I study.15) Targeting GBA, a glucosylceramide synthetase (GCS) inhibitor that prevents lipid accumulation due to decreased GBA activity is currently being tested in a Phase II study.16) A drug that enhances the GBA activity is also under development. In addition, israpidine, the aforementioned L-type calcium channel inhibitor used as an antihypertensive, completed a Phase III study for the DMT of PD.17) While all these drugs are aimed at virtually untreated early PD, substantial progression of Lewy body pathology and a 50% or more decrease in the number of dopaminergic neurons are observed at diagnosis of PD, requiring the development of biomarkers that identify patients in a very early stage or individuals at high risk of developing PD in future.
Attempts to develop early biomarkers in preclinical cohort
Idiopathic REM sleep behavior disorder (iRBD) has recently been found to be a strong risk factor for developing PD in future. With a 5-year incidence of 30%−40% and a 10-year incidence of 70%−80%, patients with iRBD may be followed up to understand the natural course of preclinical PD and identify biomarkers. In Japan, a group led by Miho Murata, National Center of Neurology and Psychiatry, is conducting a prospective cohort study to follow up approximately 100 patients with iRBD. Such cohort groups may be a good target for DMT in future.
Conclusion
The pathogenesis of PD has been elucidated through analyses of familial PD-related genes or risk genes such as rare variants and SNPs as well as recent studies based on the prion hypothesis. Many drugs have been developed accordingly, and shown to be effective in animal models as DMT; however, none can inhibit the progression of PD in humans. It may be important to further elucidate the pathogenesis and develop more appropriate animal models and biomarkers for the very early or preclinical PD.
References
- Braak, H. et al . : Cell Tissue Res ., 318, 121-134(2004)
- Schapira, A. H. V. et al . : Nat. Rev. Neurosci ., 18, 435-450(2017)
- Singleton, A. B. et al . : Science , 302, 841(2003)
- Ibáñez, P. et al . : Lancet , 364, 1169-71(2004)
- Chartier-Harlin, M. C. et al . : Lancet , 364, 1167-9(2004)
- Satake, W. et al . : Nat. Genet ., 41, 1303-7(2009)
- Simón-Sánchez, J. et al . : Nat. Genet ., 41, 1308-12(2009)
- Sidransky, E. et al . : N. Engl. J. Med ., 361, 1651-61(2009)
- Mitsui, J. et al . : Arch. Neurol ., 66, 571-6(2009)
- Mazzulli, J. R. et al . : Cell , 146, 37-52(2011)
- Luk, K. C. et al . : J. Exp. Med ., 209, 975-86(2012)
- Surmeier, D. J. et al . : Exp. Neurol ., 298, 202-209(2017)
- Lee, J. S. et al . : J. Mov. Disord ., 9, 14-9(2016)
- ClinicalTrials.gov Identifier : NCT03100149
- ClinicalTrials.gov Identifier : NCT01568099
- ClinicalTrials.gov Identifier : NCT02906020
- ClinicalTrials.gov Identifier : NCT02168842