Mass Spectrometry Reagents
Fujifilm Wako's product lineup includes protein digestive enzymes for protein mass spectrometry (MS), stable isotope-labeled amino acids for metabolomics and proteomics research, and calibration standards for MALDI-MS. The use of Lysyl Endopeptidase, which specifically cleaves the peptide bond on the carboxyl side of lysine residue (Lys-X), is becoming more common, and has been cited in over 2,500 publications.
More Information
Applications of Mass Spectrometry
In mass spectrometry (MS), components (compounds and atoms) in a sample (solids, liquids, and gases) are ionized by various ionization methods, and the mass (molecular and atomic weight) and amount of the generated ions are measured to identify the components and their ratios in the sample. A wide range of substances can be analyzed, from inorganic to organic substances of low to medium mass, and even biological molecules such as nucleic acids, sugars, peptides, and proteins. MS is used in various fields, from research in chemistry, biochemistry, and medical science to analysis of food, drug, biological samples, industrial materials, and environmental samples. With these features, MS is a powerful tool in the “omics” technologies, which have been the focus of extensive research in recent years. MS has become an indispensable analytical method for proteomics (comprehensive analysis of proteins) and metabolomics (comprehensive analysis of metabolites), including lipidomics (analysis of lipids).
Principle of Mass Spectrometry
MS technology consists of three major components. The first is the ionization of the analyte. To date, various ionization methods have been developed, including the EI and CI methods for analyzing mainly low molecular weight compounds and volatile substances, and the ESI, APCI, and MALDI methods for nonvolatile substances and biological macromolecular compounds. The second is the separation by mass, which separates the generated ions by mass (more precisely, by the mass-to-charge ratio). Separation techniques include quadrupole mass filtering, time-of-flight separation, and ion cyclotron resonance. An appropriate technique is selected based on the requirements for resolution and sensitivity. The third is the detection of the separated ions. MS is also compatible with hyphenated techniques; it is often combined with other analytical techniques such as GC, LC and ICP, to achieve synergistic effects of different analytical technologies.
Sample Pretreatment Techniques
MS involves detection of analytes based on mass, and various pretreatment techniques are applied to samples to facilitate mass separation and enhance detection of analytes. In addition, analysis of macromolecular compounds such as proteins in their native form can cause problems, such as reduced detection sensitivity or formation of multiply charged ions, which complicates the analysis of mass spectra. To solve this problem, proteins are digested into peptide fragments using enzymes in advance. The MS data of the resulting peptide mixture is compared to a protein (peptide) database for highly efficient protein identification. Below are some of the pretreatment techniques performed on samples for MS analysis.
1. Digestion of Proteins by Digestive Enzymes
As mentioned above, when the sample is a protein, it is digested by a digestive enzyme appropriate for the analyte before subjected to MS, and the resulting peptide mixture is used as the loading sample. If the digestion efficiency of the sample is low, the resulting peptide species and quantity are also low, and therefore, high quality mass spectra cannot be obtained. The enzyme trypsin is usually used for protein MS. Trypsin is the enzyme of first choice because digestion occurs specifically and efficiently at cleavage sites. Even so, digestion may become incomplete for some proteins. To circumvent the problem, Lysyl endopeptidase can be used in combination with trypsin. Like trypsin, Lysyl endopeptidase specifically and efficiently digests proteins at its cleavage sites. Using lysis endopeptidase in combination with trypsin ensures complete cleavage at lysine residues and increases the number of resulting peptides compared to using trypsin alone, thereby yielding sufficient peptide species and amounts for better detection. Good MS results facilitate protein (peptide) database searches, leading to highly accurate protein identification.
2. Quantitative Analysis with the Use of Stable Isotope-labeled Compounds
When stable isotope-labeled low-molecular-weight compounds are mixed with their unlabeled counterparts in advance and the mixture is subjected to MS, the masses of the labeled compounds differ from those of the unlabeled compounds by the mass of their isotopes, allowing quantitative analysis of the target analytes based on the detection ratio of each compound. In addition, if stable isotope-labeled low-molecular-weight compounds of known concentration are pre-mixed with unlabeled low-molecular-weight analytes, and LC/MS is performed using this mixture, the ions from both are eluted and detected at virtually the same retention time in LC/MS, so the measurement conditions (environment) are the same for both ions. As a result, absolute quantitative analysis that is unaffected by the analysis environment is possible. This method is also useful for quantitative analysis using the SRM (MRM) method, which is suitable for samples containing mixtures of multiple analytes.
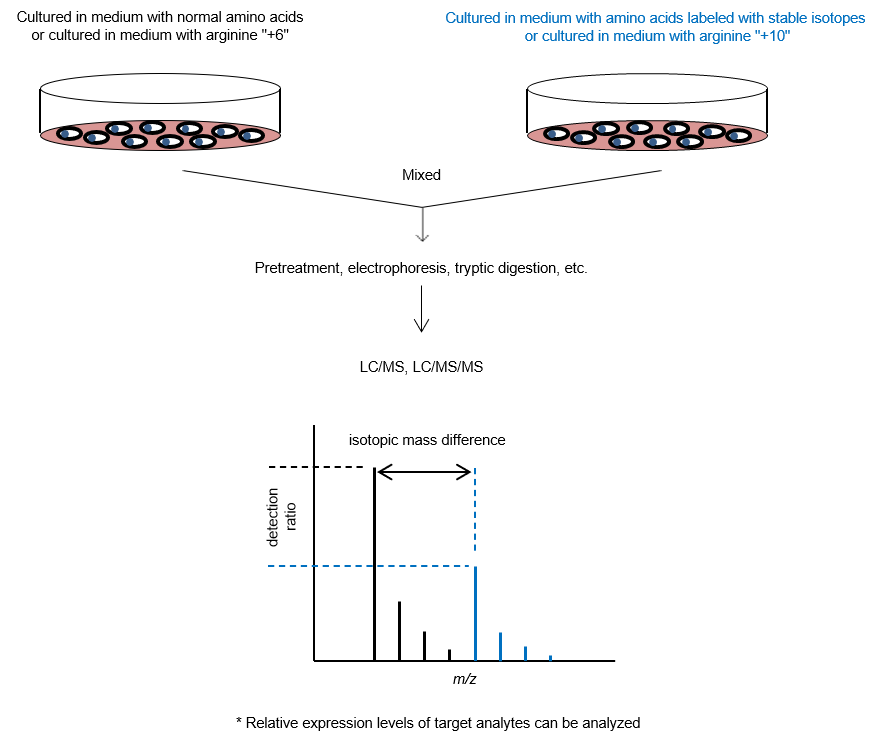
3. SILAC Method
The stable isotope labeling by amino acids in cell culture (SILAC) method1) is used as a pretreatment experiment for protein quantification. Amino acids labeled with stable isotopes (13C, 15N, etc.) are used in the cell culture medium of one of the cultures being compared, while unlabeled amino acids (normal amino acids) are used in the other culture. Cells are cultured for a certain period of time, and proteins are extracted from both cultures and mixed. The peptides after enzymatic digestion are then measured by MS to identify the proteins and quantify their expression lev els. This technique allows comparative studies of protein expression levels in cells under various culture conditions and enables comprehensive quantitative analysis of thousands of proteins. Imami et al. reported a method in which both cultures to be compared are mixed with normal amino acids, and then each culture is mixed with stable isotope-labeled arginine samples with different masses (arginine "+6" and arginine "+10")2)。
Mass Calibration in Mass Spectrometry
MS can identify and analyze the abundance of various substances by measuring their masses. To obtain reliable data, the accuracy of the dete cted mass needs be ensured. Therefore, mass calibration of the mass spectrometer must be performed in advance using standard compounds, called “calibrants,” with known mass.
Conclusions
This article has described the analysis of biological samples using mass spectrometry (MS) and the need for mass calibration. The analytical targets of MS have expanded dramatically in recent years, from small molecules to macromolecular compounds such as proteins, making MS extremely important. The technology will be further developed in the future for both qualitative and quantitative analyses.
References
- Ong, S.-E. et al.: Mol. Cell. Proteomics, 1(5), 376 (2002).
Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics - Imami, K. et al.,: Mol. Biosyst., 6(3), 594 (2010).
Quantitative proteome and phosphoproteome analyses of cultured cells based on SILAC labeling without requirement of serum dialysis
For research use or further manufacturing use only. Not for use in diagnostic procedures.
Product content may differ from the actual image due to minor specification changes etc.
If the revision of product standards and packaging standards has been made, there is a case where the actual product specifications and images are different.
The prices are list prices in Japan.Please contact your local distributor for your retail price in your region.